Spectrum of Genetic Variants Associated with Anterior Segment Dysgenesis in South Florida

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Genetic Screening
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gould, D.B.; John, S.W. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum. Mol. Genet. 2002, 11, 1185–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.A.; Walter, M.A. Genomics and anterior segment dysgenesis: A review. Clin. Exp. Ophthalmol. 2014, 42, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Strungaru, M.H.; Dinu, I.; Walter, M.A. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnicka, A.R.; Mt-Isa, S.; Owen, C.G.; Cook, D.G.; Ashby, D. Variations in primary open-angle glaucoma prevalence by age, gender, and race: A Bayesian meta-analysis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4254–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bademci, G.; Foster, J., 2nd; Mahdieh, N.; Bonyadi, M.; Duman, D.; Cengiz, F.B.; Menendez, I.; Diaz-Horta, O.; Shirkavand, A.; Zeinali, S.; et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet. Med. 2016, 18, 364–371. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Shearer, A.E.; Eppsteiner, R.W.; Booth, K.T.; Ephraim, S.S.; Gurrola, J., 2nd; Simpson, A.; Black-Ziegelbein, E.A.; Joshi, S.; Ravi, H.; Giuffre, A.C.; et al. Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am. J. Hum. Genet. 2014, 95, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Program, N.C.S.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bademci, G.; Diaz-Horta, O.; Guo, S.; Duman, D.; Van Booven, D.; Foster, J., 2nd; Cengiz, F.B.; Blanton, S.; Tekin, M. Identification of copy number variants through whole-exome sequencing in autosomal recessive nonsyndromic hearing loss. Genet. Test. Mol. Biomark. 2014, 18, 658–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Project, N.E.S.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2019, 22, 336–344. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jiang, H.; Tyler-Smith, C.; Xue, Y.; Jiang, T.; Wang, J.; Wu, M.; Liu, X.; Tian, G.; Wang, J.; et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011, 12, R95. [Google Scholar] [CrossRef] [Green Version]
- Aradhya, S.; Lewis, R.; Bonaga, T.; Nwokekeh, N.; Stafford, A.; Boggs, B.; Hruska, K.; Smaoui, N.; Compton, J.G.; Richard, G.; et al. Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet. Med. 2012, 14, 594–603. [Google Scholar] [CrossRef] [Green Version]
- D’Haene, B.; Meire, F.; Claerhout, I.; Kroes, H.Y.; Plomp, A.; Arens, Y.H.; de Ravel, T.; Casteels, I.; De Jaegere, S.; Hooghe, S.; et al. Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Investig. Ophthalmol. Vis. Sci. 2011, 52, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Ianakiev, P.; Kilpatrick, M.W.; Toudjarska, I.; Basel, D.; Beighton, P.; Tsipouras, P. Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am. J. Hum. Genet. 2000, 67, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnik Oberstein, S.A.; Kriek, M.; White, S.J.; Kalf, M.E.; Szuhai, K.; den Dunnen, J.T.; Breuning, M.H.; Hennekam, R.C. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet. 2006, 79, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weh, E.; Reis, L.M.; Happ, H.C.; Levin, A.V.; Wheeler, P.G.; David, K.L.; Carney, E.; Angle, B.; Hauser, N.; Semina, E.V. Whole exome sequence analysis of Peters anomaly. Hum. Genet. 2014, 133, 1497–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paznekas, W.A.; Boyadjiev, S.A.; Shapiro, R.E.; Daniels, O.; Wollnik, B.; Keegan, C.E.; Innis, J.W.; Dinulos, M.B.; Christian, C.; Hannibal, M.C.; et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 2003, 72, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Belmouden, A.; Melki, R.; Hamdani, M.; Zaghloul, K.; Amraoui, A.; Nadifi, S.; Akhayat, O.; Garchon, H.J. A novel frameshift founder mutation in the cytochrome P450 1B1 (CYP1B1) gene is associated with primary congenital glaucoma in Morocco. Clin. Genet. 2002, 62, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Quiroz-Casian, N.; Chacon-Camacho, O.F.; Barragan-Arevalo, T.; Nava-Valdez, J.; Lieberman, E.; Salgado-Medina, A.; Navas, A.; Graue-Hernandez, E.O.; Zenteno, J.C. Sclerocornea-Microphthalmia-Aphakia Complex: Description of Two Additional Cases Associated With Novel FOXE3 Mutations and Review of the Literature. Cornea 2018, 37, 1178–1181. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, X.; Jia, X.; Li, S.; Li, M.; Guo, X.; Liu, X.; Zhang, Q. Mutation analysis of the genes associated with anterior segment dysgenesis, microcornea and microphthalmia in 257 patients with glaucoma. Int. J. Mol. Med. 2015, 36, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Choi, A.; Lao, R.; Ling-Fung Tang, P.; Wan, E.; Mayer, W.; Bardakjian, T.; Shaw, G.M.; Kwok, P.Y.; Schneider, A.; Slavotinek, A. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur. J. Hum. Genet. 2015, 23, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.; Rudkin, A.; Parry, D.A.; Burdon, K.P.; McKibbin, M.; Logan, C.V.; Abdelhamed, Z.I.; Muecke, J.S.; Fernandez-Fuentes, N.; Laurie, K.J.; et al. Homozygous mutations in PXDN cause congenital cataract, corneal opacity, and developmental glaucoma. Am. J. Hum. Genet. 2011, 89, 464–473. [Google Scholar] [CrossRef]
- Bougeard, G.; Hadj-Rabia, S.; Faivre, L.; Sarafan-Vasseur, N.; Frebourg, T. The Rapp-Hodgkin syndrome results from mutations of the TP63 gene. Eur. J. Hum. Genet. 2003, 11, 700–704. [Google Scholar] [CrossRef]
- Salinas, C.F.; Montes, G.M. Rapp-Hodgkin syndrome: Observations on ten cases and characteristic hair changes (pili canaliculi). Birth Defects Orig. Artic. Ser. 1988, 24, 149–168. [Google Scholar] [PubMed]
- Chatterjee, M.; Neema, S.; Mukherjee, S. Rapp Hodgkin Syndrome. Indian Dermatol. Online J. 2017, 8, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.; Loidi, L.; Abalo-Lojo, J.M. Novel variant in the TP63 gene associated to ankyloblepharon-ectodermal dysplasia-cleft lip/palate (AEC) syndrome. Ophthalmic Genet. 2017, 38, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Sutton, V.R.; van Bokhoven, H. TP63-Related Disorders. In GeneReviews ((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Sutton, V.R.; Plunkett, K.; Dang, D.X.; Lewis, R.A.; Bree, A.F.; Bacino, C.A. Craniofacial and anthropometric phenotype in ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (Hay-Wells syndrome) in a cohort of 17 patients. Am. J. Med. Genet. A 2009, 149, 1916–1921. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.; Avdic, A.; Roos, B.R.; DeLuca, A.; Miller, K.; Schnieders, M.J.; Scheetz, T.E.; Alward, W.L.; Fingert, J.H. LADD syndrome with glaucoma is caused by a novel gene. Mol. Vis. 2017, 23, 179–184. [Google Scholar] [PubMed]
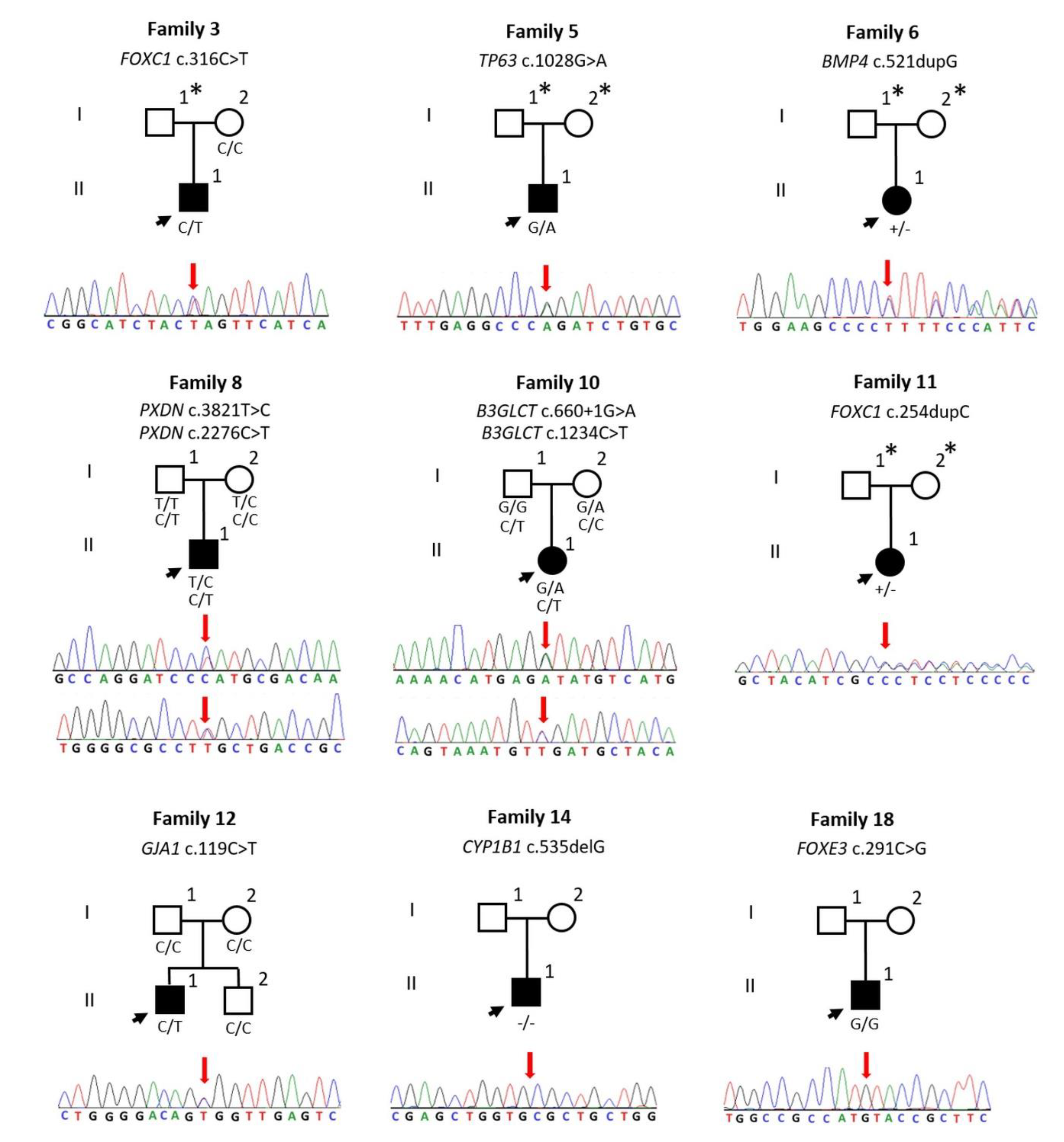

{kind=link}
| Family ID | Gene | Transcript | Inh | Zyg | cDNA | Amino Acid Change | gnomAD | CADD | GERP RS | MutationTaster | SIFT | ACMG | ACMG Guidelines | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PAX6 | NM_000280.4 | AD | HT | Large deletion (~266,752 bp) | Large deletion | N/A | N/A | N/A | N/A | N/A | LP | PVS1 | Aradhya, 2012 [19] |
| 3 | FOXC1 | NM_001453.2 | AD | HT | c.316C>T | p.Q106* | N/A | 38 | 3.8599 | DC | N/A | P | PVS1, PM2, PP3, PP5 | Dhaene, 2011 [20] |
| 5 | TP63 | NM_003722.4 | AD | HT | c.1028G>A | p.R343Q | N/A | 33 | 5.8299 | DC | DM | LP | PS3, PM2, PM5, PP3 | Ianakiev, 2000 [21] |
| 6 | BMP4 | NM_130851.3 | AD | HT | c.521dupG | p.F175Lfs*8 | N/A | 35 | 5.1999 | DC | N/A | P | PVS1, PM2, PP3 | This study |
| 8 | PXDN | NM_012293.2 | AR | HT | c.3821T>C | p.L1274P | N/A | 25.8 | 5.4099 | DC | DM | VUS | PM2, PP3 | This study |
| HT | c.2276C>T | p.S759L | 0.00001204 | 32 | 5.63 | DC | DM | VUS | PM2, PP3, | This study | ||||
| 10 | B3GLCT | NM_194318.3 | AR | HT | c.660+1G>A | Splice | 0.0007602 | 34 | 6.0799 | DC | N/A | P | PVS1, PP3, PP5 | Lesnik Oberstein, 2006 [22] |
| HT | c.1234C>T | p.R412* | N/A | 36 | 3.23 | DC | N/A | P | PVS1, PM2, PP3, PP5 | Weh, 2014 [23] | ||||
| 11 | FOXC1 | NM_001453.2 | AD | HT | c.254dupC | p.L86Afs*220 | N/A | 33 | 0.5139 | DC | N/A | P | PVS1, PM1, PM2 | This study |
| 12 | GJA1 | NM_000165.4 | AD | HT | c.119C>T | p.A40V | N/A | 25.5 | 6.1599 | DC | DM | P | PS3, PM1, PM2, PM6, PP2, PP3, PP5 | Paznekas, 2003 [24] |
| 14 | CYP1B1 | NM_000104.3 | AR | HM | c.535delG | p.A179Rfs*18 | 0.00004797 | 24.2 | 2.5599 | DC | N/A | P | PVS1, PM2, PP5 | Belmouden, 2002 [25] |
| 18 | FOXE3 | NM_012186.2 | AR | HM | c.291C>G | p.I97M | 0.00002015 | 22.4 | 1.1799 | DC | DM | VUS | PM2, PP3 | Quiroz-Casian, 2018 [26] |
| Family-Individual ID | Sex | Simplex/Multiplex | Age (Years) | Ethnicity | Eye Phenotype | Additional Clinical Features | Gene |
|---|---|---|---|---|---|---|---|
| 1-II:1 | M | Sx | 9 | Black, non-Hispanic | Aniridia with glaucoma | - | PAX6 |
| 3-II:1 | M | Sx | 11 | White, non-Hispanic | AR with glaucoma | - | FOXC1 |
| 5-II:1 | M | Sx | 9 | White, Hispanic | Peters anomaly OD | Syndactyly of third and fourth toes in the left foot, vesicoureteral reflux, cleft lip and palate, and nasolacrimal abnormalities | TP63 |
| 6-II:1 | F | Sx | 9 | White, Hispanic | Peters anomaly OU | - | BMP4 |
| 8-II:1 | M | Sx | 8 | White, non-Hispanic | Peters anomaly OU | - | PXDN |
| 10-II:1 | F | Sx | 8 | White, Hispanic | Peters anomaly OU | - | B3GLCT |
| 11-II:1 | F | Sx | 37 | White, Hispanic | AR with glaucoma | - | FOXC1 |
| 12-II:1 | M | Sx | 13 | White, Hispanic | Microphthalmia with glaucoma | Microdontia, underdeveloped alae nasi, syndactyly | GJA1 |
| 14-II:1 | M | Sx | 13 | White, Hispanic | Peters anomaly OU | - | CYP1B1 |
| 18-II:1 | M | Sx | 6 | White, Hispanic | Peters anomaly OU | - | FOXE3 |
| Studies | Sample Size | Phenotypes | Population Studied | ES or Gene Panel | Causative Variants Detected in ASD |
|---|---|---|---|---|---|
| Weh et al. [23] | 27 | Syndromic Peters anomaly: 20 Isolated Peters anomaly: 7 | Children’s Hospital of Wisconsin (USA) Population subtypes were not mentioned | ES | 22.2% overall |
| Huang et al. [27] | 257 | POAG: 125 PACG: 132 | Chinese: 257 | ES of 43 genes associated with ASD, microcornea or microphthalmia | 10.9% overall POAG: 8.80% PACG: 12.9% |
| Patel et al. [28] | 277 | MAC: 98 cases ASDA: 113 cases Other or syndromic: 8 cases RET: 49 cases Congenital cataracts and lens-associated conditions: 9 cases | White European: 139 South Asian: 21 Black African: 7 Arabic or Middle Eastern: 5 Black Caribbean: 2 Unknown: 91 Mixed/unclassified: 12 | Oculome panel of 429 known eye disease genes | 24.5% overall Congenital cataracts and lens-associated conditions: 88.9% RET: 42.8% Other or syndromic: 37.5% ASD: 24.8% |
| This study | 24 | Peters anomaly: 8 PCG: 6 AR: 5 Aniridia: 2 Congenital corneal dystrophy: 1 Microphthalmia with glaucoma: 1 | White, Hispanic: 11 (7 solved) Black, Hispanic: 1 (0 solved) White, non-Hispanic: 6 (2 solved) Black, non-Hispanic: 6 (1 solved) | ES of 92 genes associated with eye phenotypes | 42% overall Peters anomaly: 75% Aniridia: 50% Others: 50% AR: 40% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thanikachalam, S.; Hodapp, E.; Chang, T.C.; Morel Swols, D.; Cengiz, F.B.; Guo, S.; Zafeer, M.F.; Seyhan, S.; Bademci, G.; Scott, W.K.; et al. Spectrum of Genetic Variants Associated with Anterior Segment Dysgenesis in South Florida. Genes 2020, 11, 350. https://doi.org/10.3390/genes11040350
Thanikachalam S, Hodapp E, Chang TC, Morel Swols D, Cengiz FB, Guo S, Zafeer MF, Seyhan S, Bademci G, Scott WK, et al. Spectrum of Genetic Variants Associated with Anterior Segment Dysgenesis in South Florida. Genes. 2020; 11(4):350. https://doi.org/10.3390/genes11040350
Chicago/Turabian StyleThanikachalam, Saradadevi, Elizabeth Hodapp, Ta C. Chang, Dayna Morel Swols, Filiz B. Cengiz, Shengru Guo, Mohammad F. Zafeer, Serhat Seyhan, Guney Bademci, William K. Scott, and et al. 2020. "Spectrum of Genetic Variants Associated with Anterior Segment Dysgenesis in South Florida" Genes 11, no. 4: 350. https://doi.org/10.3390/genes11040350