Extracellular Vesicle-Mediated siRNA Delivery, Protein Delivery, and CFTR Complementation in Well-Differentiated Human Airway Epithelial Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
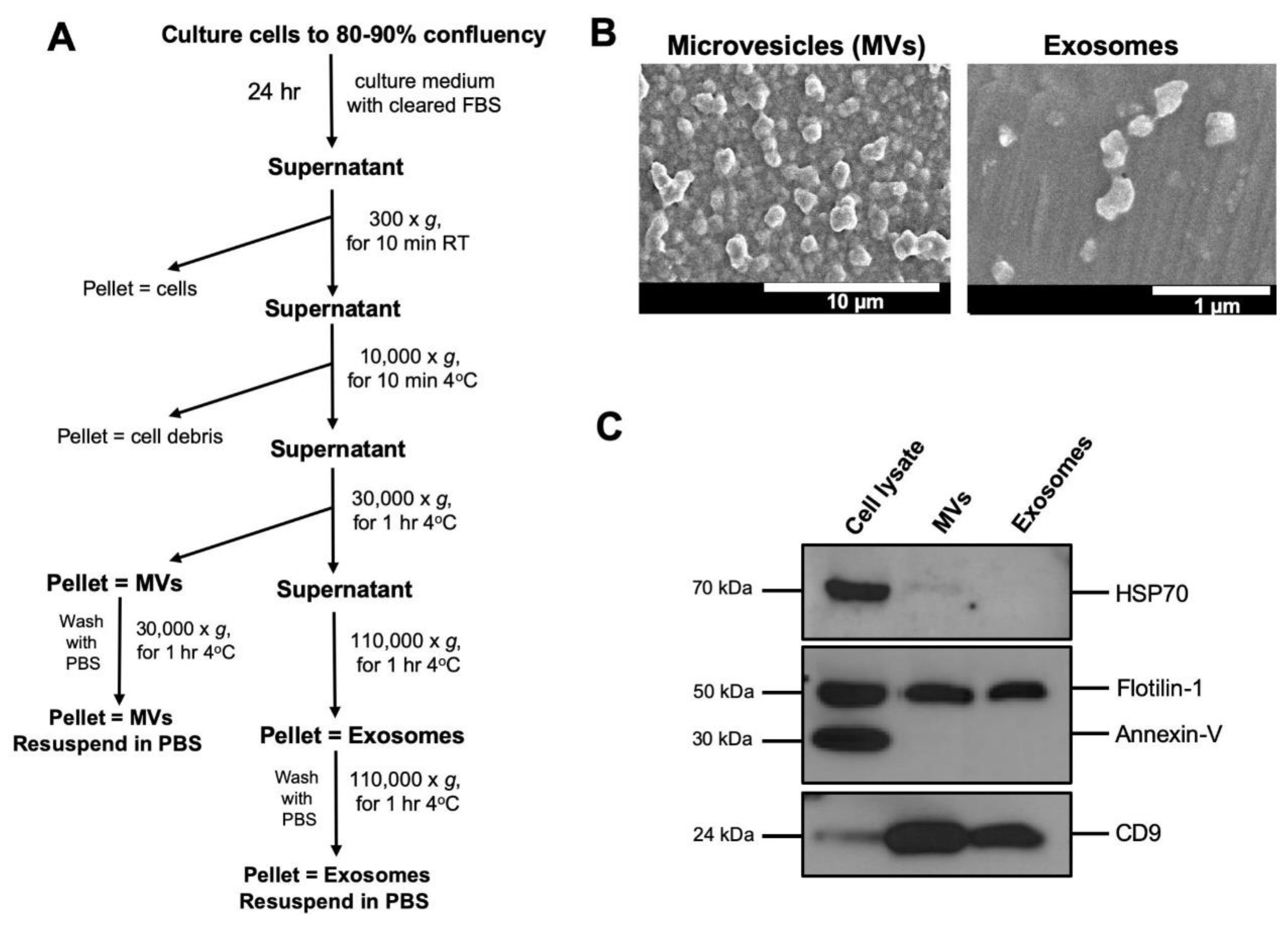
2.2. Isolation of Extracellular Vesicles (EVs)
2.3. Electron Microscopy
2.4. SDS-PAGE and Immunoblot Analysis
2.5. Dicer-Substrate Short Interfering RNA (DsiRNA) Oligonucleotides
2.6. Loading of DsiRNA into Exosomes
2.7. Exosome Labeling
2.8. Confocal Microscopy and Immunostaining
2.9. Real-Time Quantitative PCR (RT-qPCR)
2.10. Isolation of CFTR- or mCherry-Loaded EVs and Delivery to HAE
2.11. Ussing Chamber Studies of Well-Differentiated HAE
3. Results
3.1. Isolation and Characterization of Extracellular Vesicles
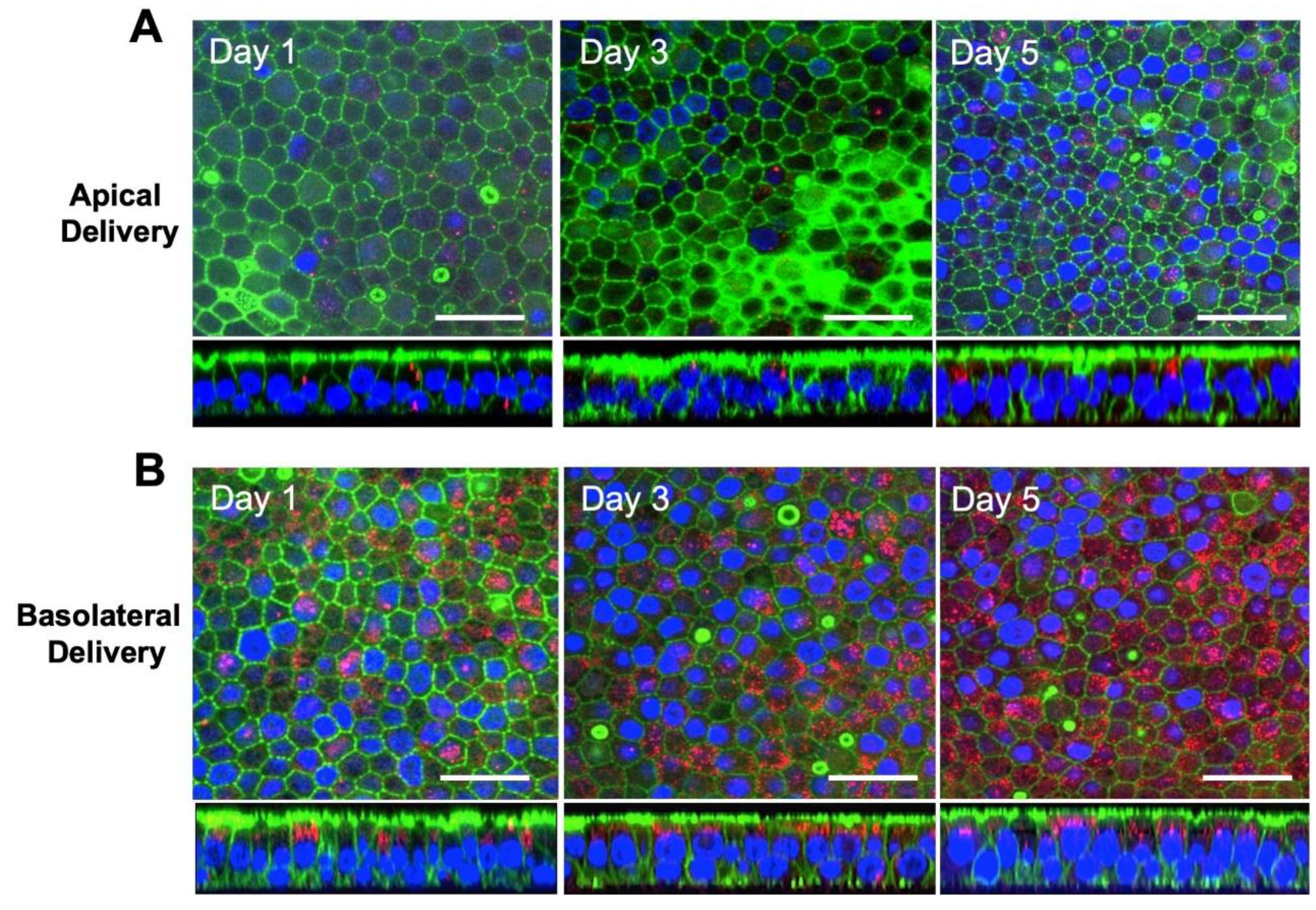
3.2. Exosomes are Internalized at the Basolateral Surface of HAE
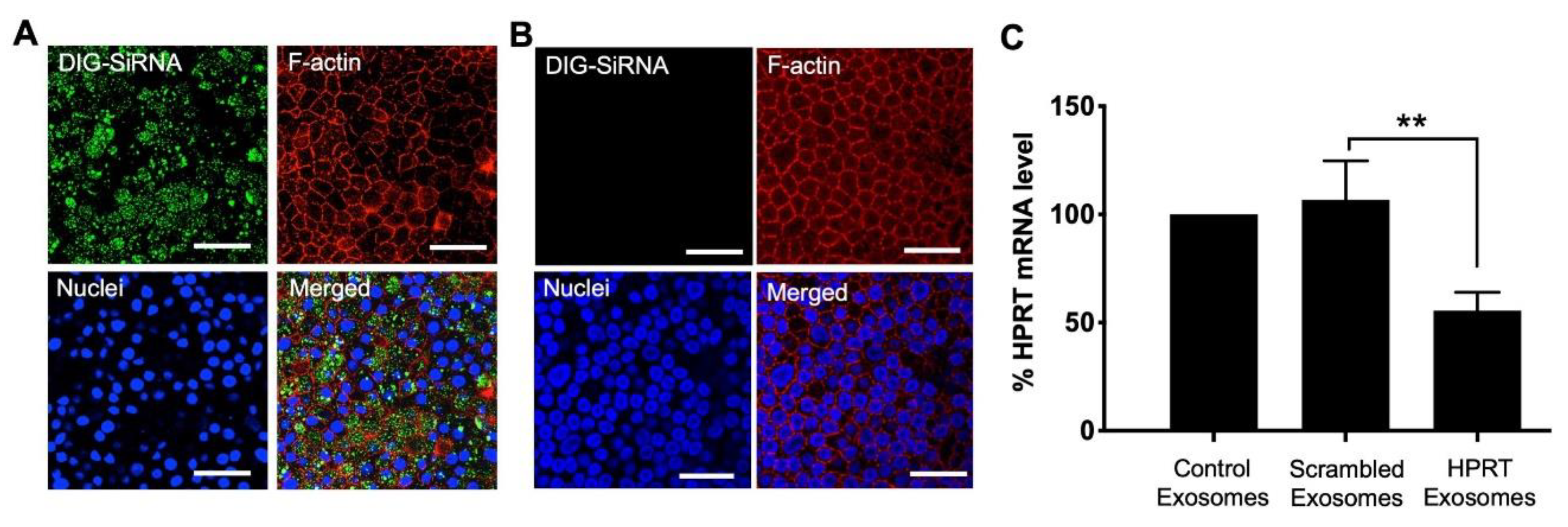
3.3. Delivery of siRNA into HAE Using Exosomes
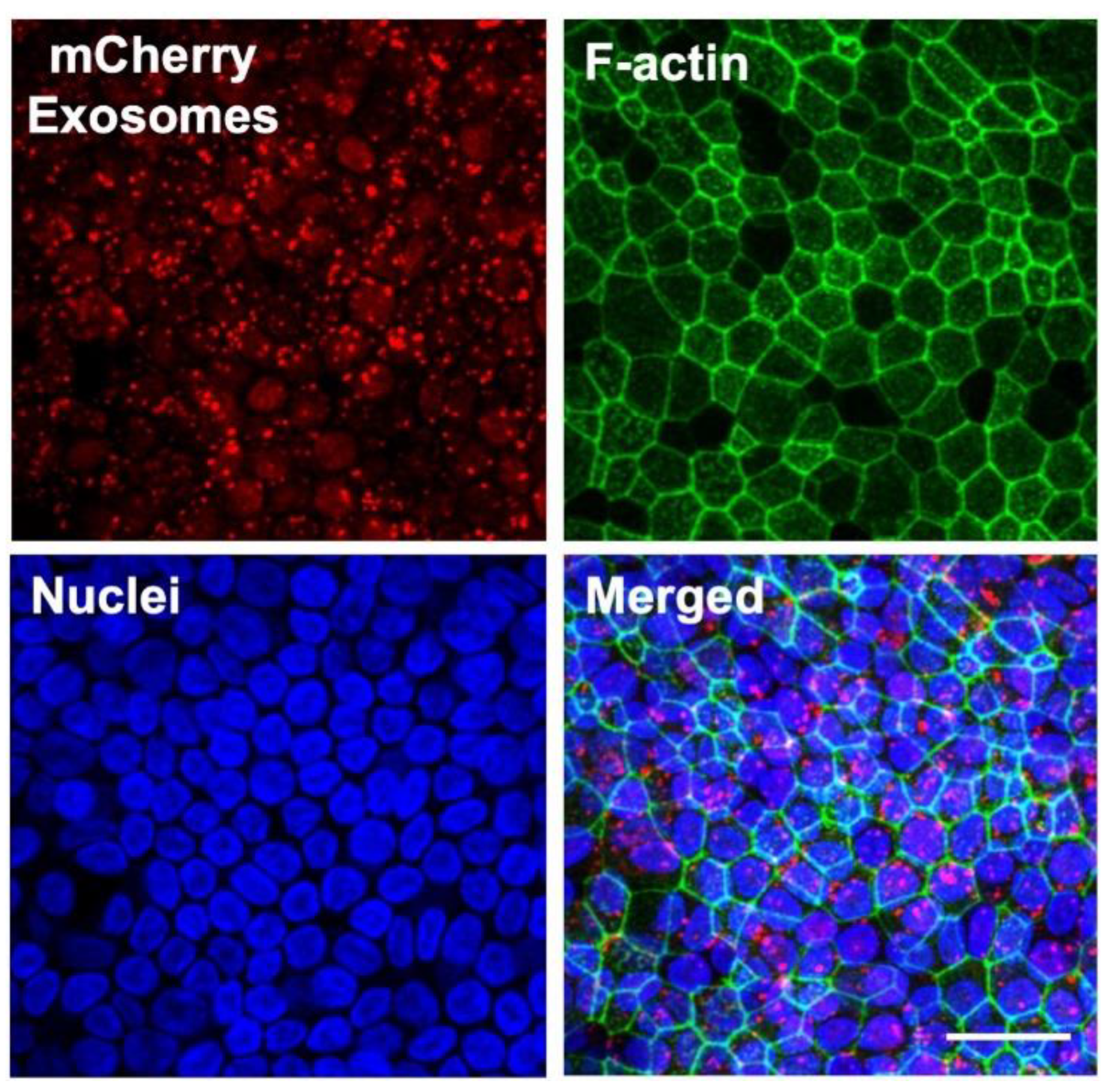
3.4. Protein Delivery to HAE Using Exosomes
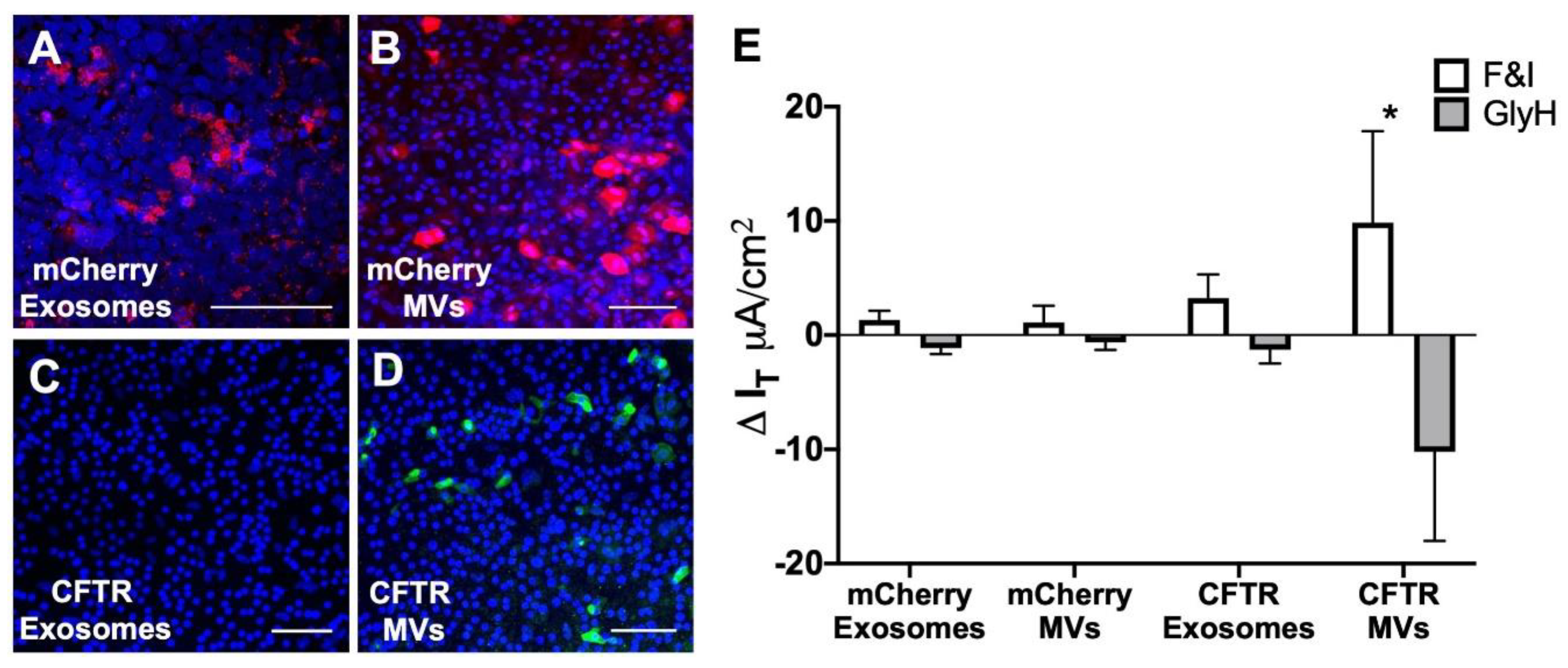
3.5. MV-Mediated Delivery of CFTR Protein Corrects Anion Defect in HAE from CF Donors
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005, 107, 183–206. [Google Scholar] [PubMed]
- Karp, P.H.; Moninger, T.O.; Weber, S.P.; Nesselhauf, T.S.; Launspach, J.L.; Zabner, J.; Welsh, M.J. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol. Biol. 2002, 188, 115–137. [Google Scholar] [PubMed]
- Woodworth, B.A.; Antunes, M.B.; Bhargave, G.; Palmer, J.N.; Cohen, N.A. Murine tracheal and nasal septal epithelium for air-liquid interface cultures: A comparative study. Am. J. Rhinol. 2007, 21, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Zeiher, B.G.; Friedman, E.; Welsh, M.J. Adenovirus-mediated gene transfer to ciliated airway epithelia requires prolonged incubation time. J. Virol. 1996, 70, 6994–7003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caci, E.; Melani, R.; Pedemonte, N.; Yueksekdag, G.; Ravazzolo, R.; Rosenecker, J.; Galietta, L.J.; Zegarra-Moran, O. Epithelial sodium channel inhibition in primary human bronchial epithelia by transfected sirna. Am. J. Respir. Cell Mol. Biol. 2009, 40, 211–216. [Google Scholar] [CrossRef]
- Griesenbach, U.; Kitson, C.; Escudero Garcia, S.; Farley, R.; Singh, C.; Somerton, L.; Painter, H.; Smith, R.L.; Gill, D.R.; Hyde, S.C.; et al. Inefficient cationic lipid-mediated sirna and antisense oligonucleotide transfer to airway epithelial cells in vivo. Respir. Res. 2006, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.; Behlke, M.A.; Ramachandran, S.; Salem, A.K.; McCray, P.B., Jr.; Davidson, B.L. Manipulation of cell physiology enables gene silencing in well-differentiated airway epithelia. Mol. Ther. Nucleic Acids 2012, 1, e41. [Google Scholar] [CrossRef]
- Platz, J.; Pinkenburg, O.; Beisswenger, C.; Puchner, A.; Damm, T.; Bals, R. Application of small interfering RNA (sirna) for modulation of airway epithelial gene expression. Oligonucleotides 2005, 15, 132–138. [Google Scholar] [CrossRef]
- Ramachandran, S.; Krishnamurthy, S.; Jacobi, A.M.; Wohlford-Lenane, C.; Behlke, M.A.; Davidson, B.L.; McCray, P.B., Jr. Efficient delivery of RNA interference oligonucleotides to polarized airway epithelia in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L23–L32. [Google Scholar] [CrossRef] [Green Version]
- Hutt, D.M.; Herman, D.; Rodrigues, A.P.; Noel, S.; Pilewski, J.M.; Matteson, J.; Hoch, B.; Kellner, W.; Kelly, J.W.; Schmidt, A.; et al. Reduced histone deacetylase 7 activity restores function to misfolded cftr in cystic fibrosis. Nat. Chem. Biol. 2010, 6, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The hsc70 co-chaperone chip targets immature cftr for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Barriere, H.; Bagdany, M.; Rabeh, W.M.; Du, K.; Hohfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded cftr from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, N.; Lankar, D.; Faure, F.; Regnault, A.; Dumont, C.; Raposo, G.; Hivroz, C. Tcr activation of human t cells induces the production of exosomes bearing the tcr/cd3/zeta complex. J. Immunol. 2002, 168, 3235–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Thery, C.; Regnault, A.; Garin, J.; Wolfers, J.; Zitvogel, L.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 1999, 147, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef]
- Lasser, C.; Alikhani, V.S.; Ekstrom, K.; Eldh, M.; Paredes, P.T.; Bossios, A.; Sjostrand, M.; Gabrielsson, S.; Lotvall, J.; Valadi, H. Human saliva, plasma and breast milk exosomes contain RNA: Uptake by macrophages. J. Transl. Med. 2011, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular vesicles: Composition, biological relevance, and methods of study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of sirna to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, S.; Lakhal, S.; Mager, I.; Wood, M.J. Exosomes for targeted sirna delivery across biological barriers. Adv. Drug Deliv. Rev. 2013, 65, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M. The jeanne manery-fisher memorial lecture 1991. Maturation of reticulocytes: Formation of exosomes as a mechanism for shedding membrane proteins. Biochem. Cell Biol. Biochim. Et Biol. Cell. 1992, 70, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Et Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef]
- Zomer, A.; Vendrig, T.; Hopmans, E.S.; van Eijndhoven, M.; Middeldorp, J.M.; Pegtel, D.M. Exosomes: Fit to deliver small RNA. Commun. Integr. Biol. 2010, 3, 447–450. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Zhuang, X.; Zhang, S.; Deng, Z.B.; Grizzle, W.; Miller, D.; Zhang, H.G. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv. Drug Deliv. Rev. 2013, 65, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of extracellular vesicles: General methodologies and latest trends. Biomed. Res. Int. 2018, 2018, 8545347. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational sirna design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef]
- Amarzguioui, M.; Lundberg, P.; Cantin, E.; Hagstrom, J.; Behlke, M.A.; Rossi, J.J. Rational design and in vitro and in vivo delivery of dicer substrate sirna. Nat. Protoc. 2006, 1, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Collingwood, M.A.; Rose, S.D.; Huang, L.; Hillier, C.; Amarzguioui, M.; Wiiger, M.T.; Soifer, H.S.; Rossi, J.J.; Behlke, M.A. Chemical modification patterns compatible with high potency dicer-substrate small interfering rnas. Oligonucleotides 2008, 18, 187–200. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Lee, Y.; Lakhal-Littleton, S.; Li, J.; Seow, Y.; Gardiner, C.; Alvarez-Erviti, L.; Sargent, I.L.; Wood, M.J. Exosome-mediated delivery of sirna in vitro and in vivo. Nat. Protoc. 2012, 7, 2112–2126. [Google Scholar] [CrossRef]
- Cooney, A.L.; Singh, B.K.; Sinn, P.L. Hybrid nonviral/viral vector systems for improved piggybac DNA transposon in vivo delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Cooney, A.L.; Zhang, W.; Ehrhardt, A.; Sinn, P.L. Enhanced tropism of species b1 adenoviral-based vectors for primary human airway epithelial cells. Mol. Methods Clin. Dev. 2019, 14, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sheppard, D.N.; Hug, M.J. Transepithelial electrical measurements with the ussing chamber. J. Cyst. Fibros 2004, 3, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Cooney, A.L.; Abou Alaiwa, M.H.; Shah, V.S.; Bouzek, D.C.; Stroik, M.R.; Powers, L.S.; Gansemer, N.D.; Meyerholz, D.K.; Welsh, M.J.; Stoltz, D.A.; et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Stout, J.T.; Caskey, C.T. Hprt: Gene structure, expression, and mutation. Annu. Rev. Genet. 1985, 19, 127–148. [Google Scholar] [CrossRef]
- Olsen, J.C.; Johnson, L.G.; Stutts, M.J.; Sarkadi, B.; Yankaskas, J.R.; Swanstrom, R.; Boucher, R.C. Correction of the apical membrane chloride permeability defect in polarized cystic fibrosis airway epithelia following retroviral-mediated gene transfer. Hum. Gene 1992, 3, 253–266. [Google Scholar] [CrossRef]
- Cooney, A.L.; Singh, B.K.; Loza, L.M.; Thornell, I.M.; Hippee, C.E.; Powers, L.S.; Ostedgaard, L.S.; Meyerholz, D.K.; Wohlford-Lenane, C.; Stoltz, D.A.; et al. Widespread airway distribution and short-term phenotypic correction of cystic fibrosis pigs following aerosol delivery of piggybac/adenovirus. Nucleic Acids Res. 2018, 46, 9591–9600. [Google Scholar] [CrossRef] [Green Version]
- Sinn, P.L.; Cooney, A.L.; Oakland, M.; Dylla, D.E.; Wallen, T.J.; Pezzulo, A.A.; Chang, E.H.; McCray, P.B., Jr. Lentiviral vector gene transfer to porcine airways. Mol. Ther. Nucleic Acids 2012, 1, e56. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Starner, T.D.; Scheetz, T.E.; Traver, G.L.; Tilley, A.E.; Harvey, B.G.; Crystal, R.G.; McCray, P.B., Jr.; Zabner, J. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L25–L31. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, S.; Karp, P.H.; Jiang, P.; Ostedgaard, L.S.; Walz, A.E.; Fisher, J.T.; Keshavjee, S.; Lennox, K.A.; Jacobi, A.M.; Rose, S.D.; et al. A microrna network regulates expression and biosynthesis of wild-type and deltaf508 mutant cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 2012, 109, 13362–13367. [Google Scholar] [CrossRef] [Green Version]
- Toumey, C. In the footsteps of biotech. Nat. Nanotechnol. 2010, 5, 475. [Google Scholar] [CrossRef]
- Bakand, S.; Hayes, A.; Dechsakulthorn, F. Nanoparticles: A review of particle toxicology following inhalation exposure. Inhal. Toxicol. 2012, 24, 125–135. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Aggarwal, P.; Hall, J.B.; McNeil, S.E. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharm. 2008, 5, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Iavicoli, I.; Leso, V.; Fontana, L.; Bergamaschi, A. Toxicological effects of titanium dioxide nanoparticles: A review of in vitro mammalian studies. Eur. Rev. Med Pharmacol. Sci. 2011, 15, 481–508. [Google Scholar] [PubMed]
- Maynard, A.D. Don’t define nanomaterials. Nature 2011, 475, 31. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles—Endogenous nanocarriers for targeted cancer therapy. Biochim. Et Biophys. Acta 2014, 1846, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Thery, C. Exosomes: Immune properties and potential clinical implementations. Semin. Immunopathol. 2011, 33, 419–440. [Google Scholar] [CrossRef]
- Marcus, M.E.; Leonard, J.N. Fedexosomes: Engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals 2013, 6, 659–680. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.S.; Lim, S.K. Measurement of precursor mirna in exosomes from human esc-derived mesenchymal stem cells. Methods Mol. Biol. 2013, 1024, 69–86. [Google Scholar]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of functional anti-mir-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef]
- Wahlgren, J.; De, L.K.T.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Strobel, T.; Breakefield, X.O.; Saydam, O. Genetically engineered microvesicles carrying suicide mrna/protein inhibit schwannoma tumor growth. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laakkonen, P.; Vuorinen, K. Homing peptides as targeted delivery vehicles. Integr. Biol. Quant. Biosci. Nano Macro 2010, 2, 326–337. [Google Scholar] [CrossRef]
- Nakase, I.; Futaki, S. Combined treatment with a ph-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep. 2015, 5, 10112. [Google Scholar] [CrossRef]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef] [Green Version]
- Sinn, P.; Williams, G.; Vongpunsawad, S.; Cattaneo, R.; McCray, P. Measles virus preferentially transduces the basolateral surface of well-differentiated human airway epithelia. J. Virol. 2002, 76, 2403–2409. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Radicioni, G.; Abdelwahab, S.; Dang, H.; Carpenter, J.; Chua, M.; Mieczkowski, P.A.; Sheridan, J.T.; Randell, S.H.; Kesimer, M. Intercellular communication between airway epithelial cells is mediated by exosome-like vesicles. Am. J. Respir. Cell Mol. Biol. 2019, 60, 209–220. [Google Scholar] [CrossRef]
- Kesimer, M.; Scull, M.; Brighton, B.; DeMaria, G.; Burns, K.; O’Neal, W.; Pickles, R.J.; Sheehan, J.K. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: A possible role in innate defense. FASEB J. 2009, 23, 1858–1868. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DsiRNA | Target mRNA | DsiRNA Target Sequence |
|---|---|---|
| hHPRT | human HPRT | 5′ pGCCAGACUUUGUUGGAUUUGAATT |
| 3′ UCGGUCUGAAACAACCUAAACUUUAA | ||
| DIG-HPRT | human HPRT | 5′ pCCAGUAAAGUUATCACAUGUUCUAG |
| 3′ GUGGUCAUUUCAAUAGUGUACAAGAUC | ||
| Scrambled | Not applicable | 5′ pCGUUAAUCGCGUAUAAUACGCGUAT |
| 3′ CAGCAAUUAGCGCAUAUUAUGCCGCAUA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, B.K.; Cooney, A.L.; Krishnamurthy, S.; Sinn, P.L. Extracellular Vesicle-Mediated siRNA Delivery, Protein Delivery, and CFTR Complementation in Well-Differentiated Human Airway Epithelial Cells. Genes 2020, 11, 351. https://doi.org/10.3390/genes11040351
Singh BK, Cooney AL, Krishnamurthy S, Sinn PL. Extracellular Vesicle-Mediated siRNA Delivery, Protein Delivery, and CFTR Complementation in Well-Differentiated Human Airway Epithelial Cells. Genes. 2020; 11(4):351. https://doi.org/10.3390/genes11040351
Chicago/Turabian StyleSingh, Brajesh K., Ashley L. Cooney, Sateesh Krishnamurthy, and Patrick L. Sinn. 2020. "Extracellular Vesicle-Mediated siRNA Delivery, Protein Delivery, and CFTR Complementation in Well-Differentiated Human Airway Epithelial Cells" Genes 11, no. 4: 351. https://doi.org/10.3390/genes11040351