Knockout of IRF7 Highlights its Modulator Function of Host Response Against Avian Influenza Virus and the Involvement of MAPK and TOR Signaling Pathways in Chicken



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
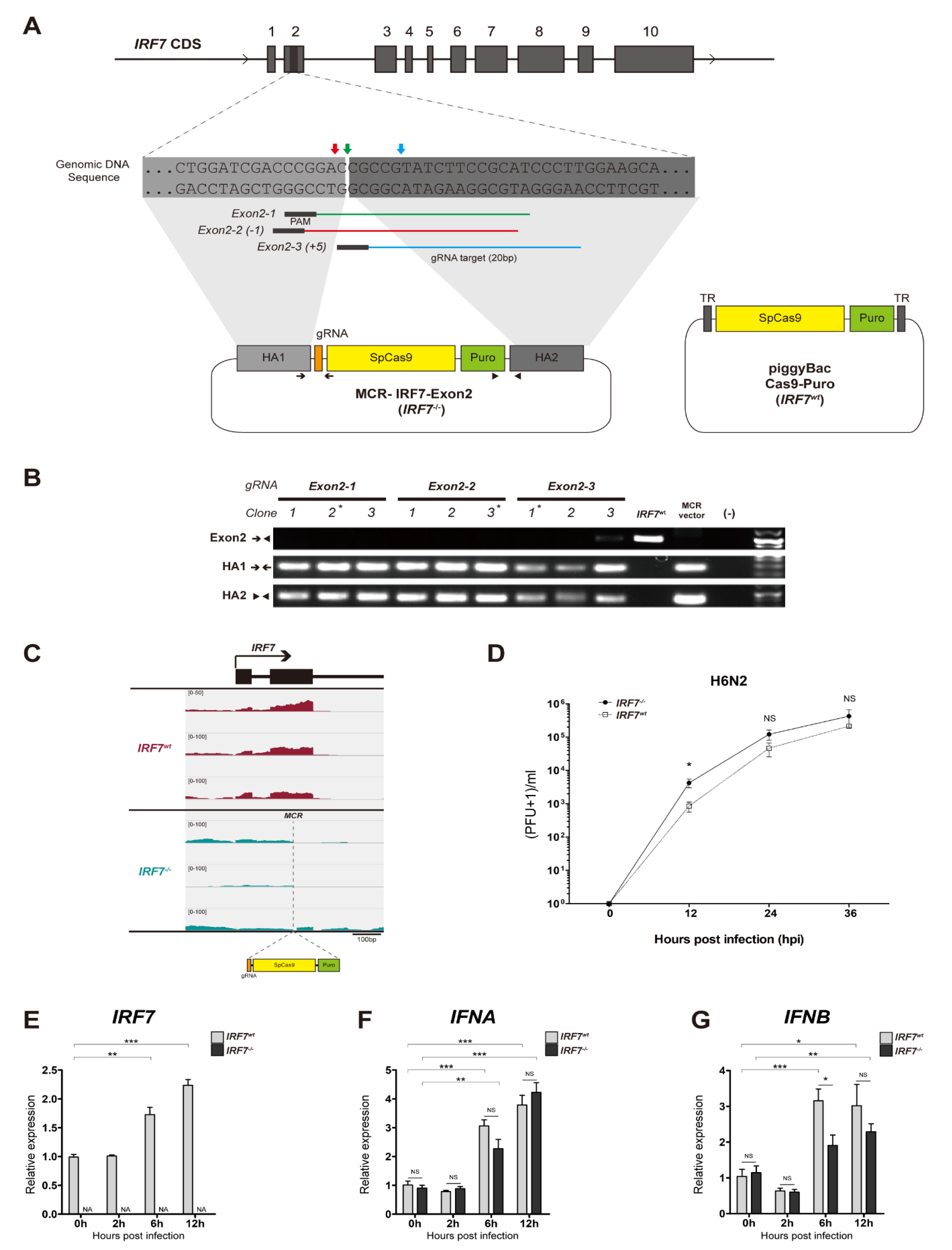
2.1. Construction of IRF7 Knockout MCR Plasmids
2.2. IRF7 Knockout Cell Clone Generation
2.3. IRF7 Knockout Clone Genotyping
2.4. Cas9-Puro Cell Line Generation
2.5. Quantitative Reverse Transcriptase PCR
2.6. AIV and In Vitro Infection
2.7. RNA Sequencing and Data Analysis
3. Results
3.1. Establishment of IRF7 Knockout DF-1 Cell Clones
3.2. LPAIV In Vitro Infection
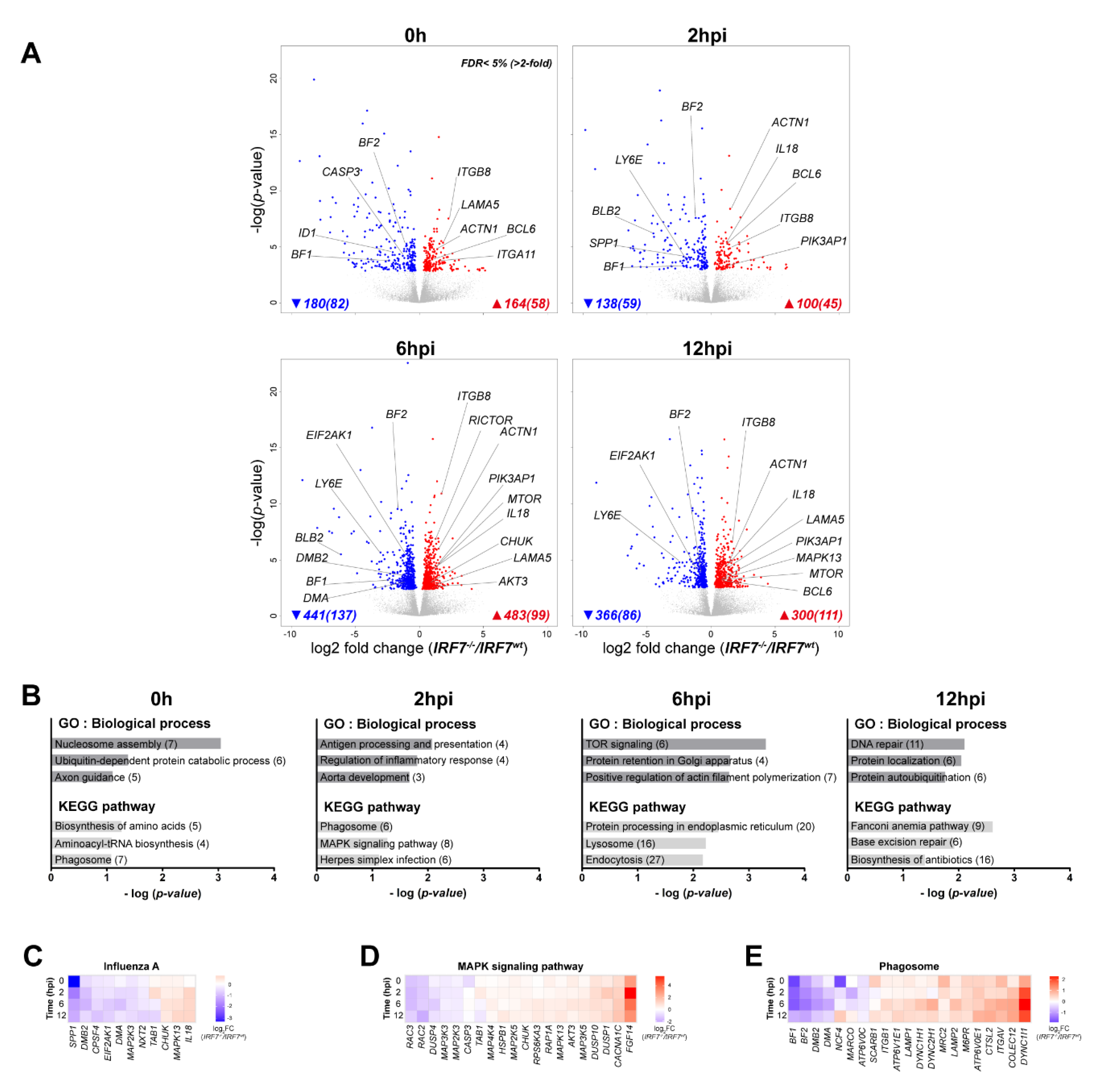
3.3. The Effect of IRF7 Knockout on the Host Response Against H6N2 Infection
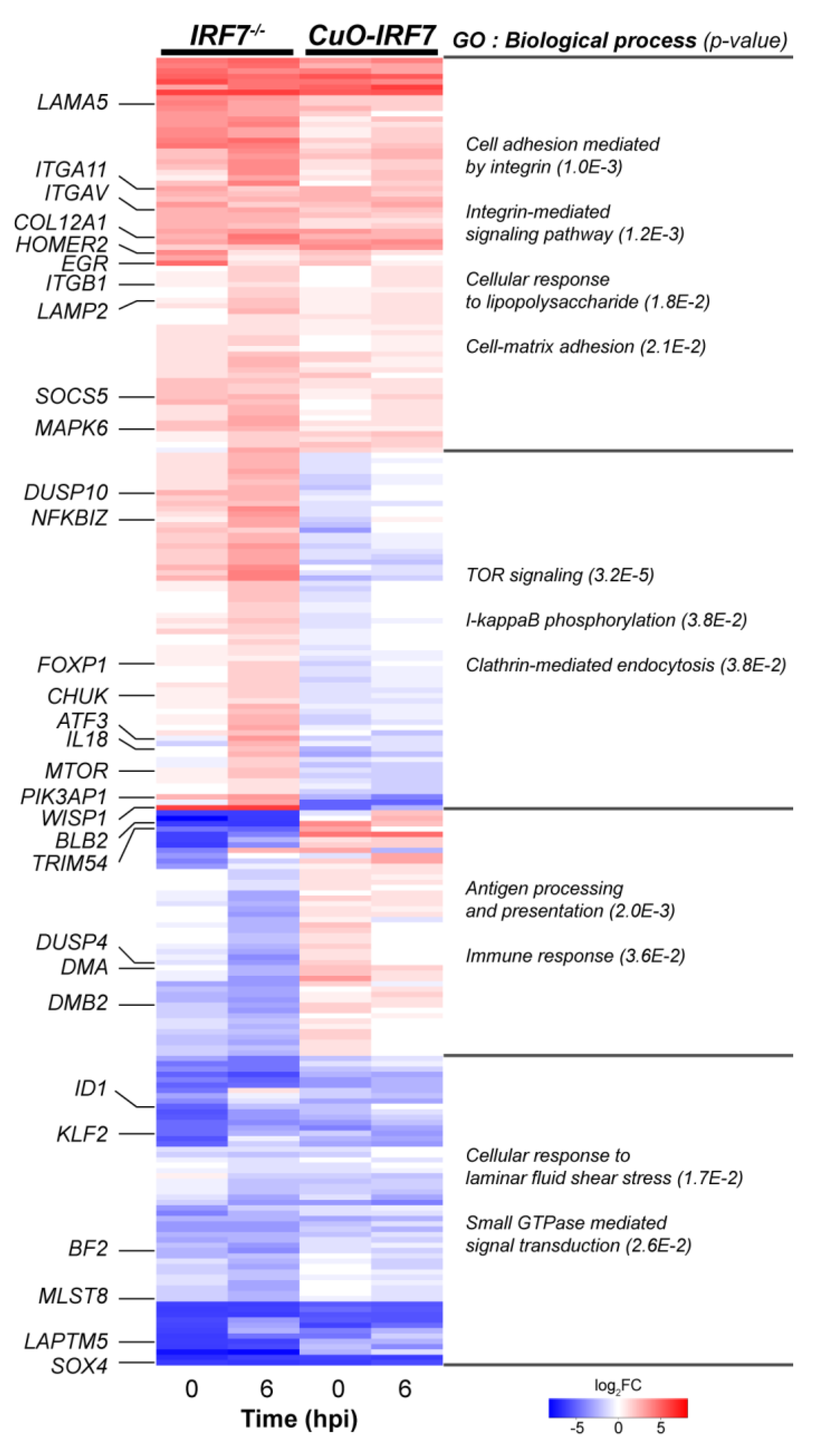
3.4. Comparative Analysis of DEGs Between IRF7 Knockout and Overexpression
3.5. Differential Expression Analysis of the Host Response upon H6N2 Infection
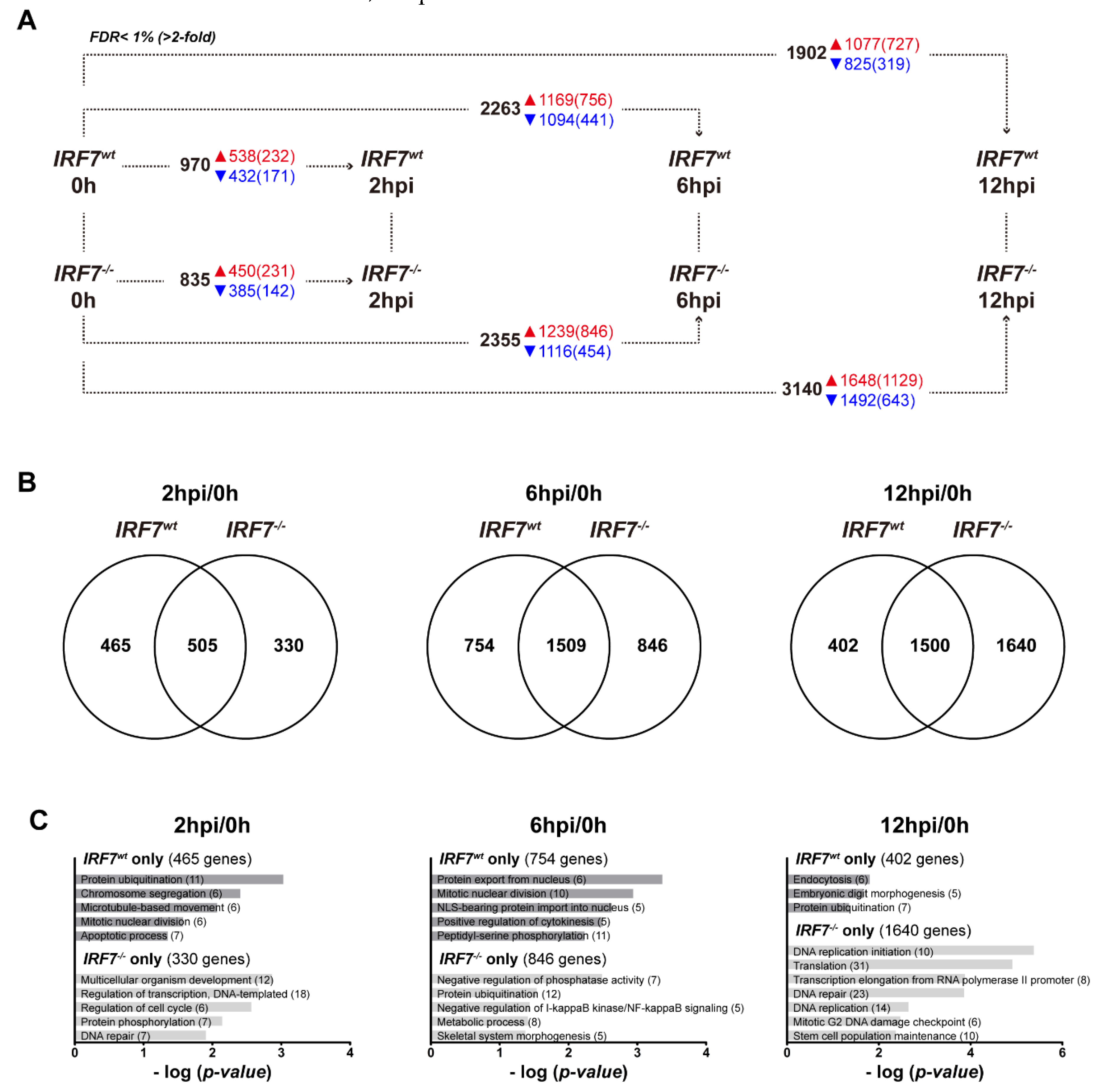
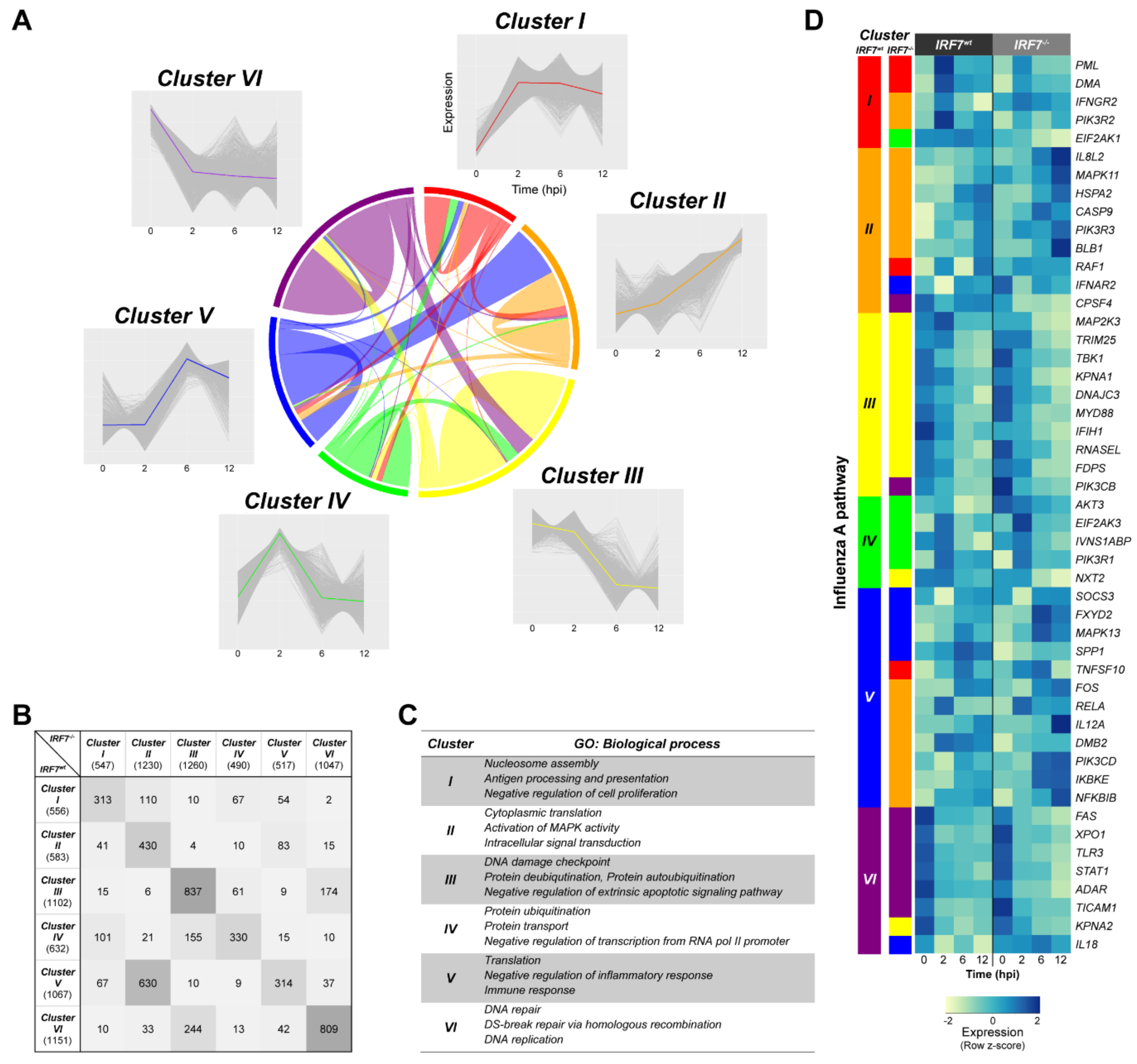
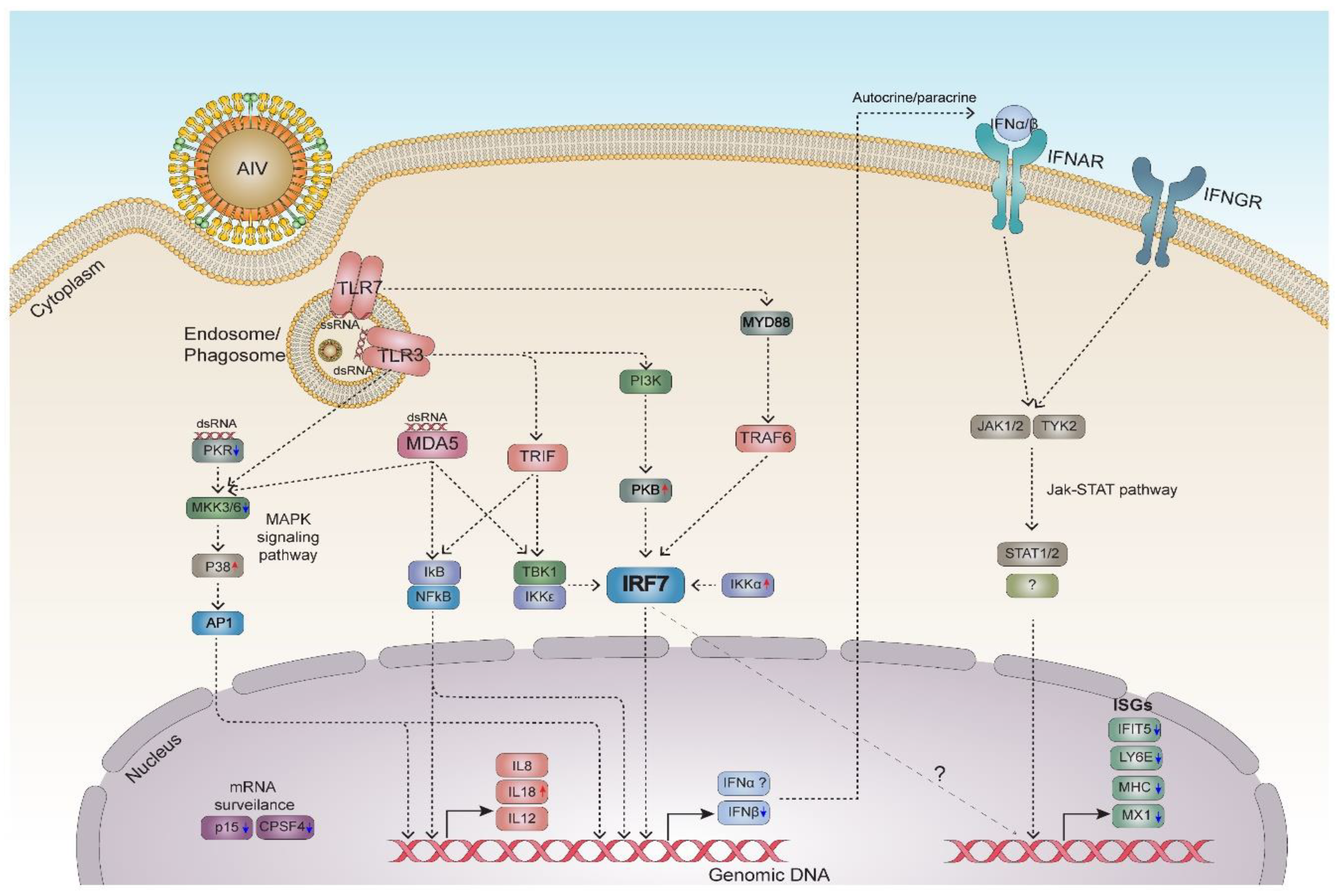
3.6. Dynamic Gene Expression Profile Analysis Revealed an Alternative Pathway to IRF7 Regulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenstrom, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza a virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swayne, D.E. Impact of vaccines and vaccination on global control of avian influenza. Avian Dis. 2012, 56, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Jhung, M.A.; Nelson, D.I.; Centers for Disease Control and Prevention. Outbreaks of avian influenza A (H5N2), (H5N8), and (H5N1) among birds—United States, December 2014–January 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 111. [Google Scholar] [PubMed]
- Ramos, S.; MacLachlan, M.; Melton, A. Impacts of the 2014-2015 Highly Pathogenic Avian Influenza Outbreak on the U.S. Poultry Sector; US Department of Agriculture World Agriculture Outlook Board: Washington, DC, USA, 2017.
- Wang, X.; Jiang, H.; Wu, P.; Uyeki, T.M.; Feng, L.; Lai, S.; Wang, L.; Huo, X.; Xu, K.; Chen, E.; et al. Epidemiology of avian influenza A H7N9 virus in human beings across five epidemics in mainland China, 2013–2017: An epidemiological study of laboratory-confirmed case series. Lancet Infect. Dis. 2017, 17, 822–832. [Google Scholar] [CrossRef]
- Lai, S.; Qin, Y.; Cowling, B.J.; Ren, X.; Wardrop, N.A.; Gilbert, M.; Tsang, T.K.; Wu, P.; Feng, L.; Jiang, H.; et al. Global epidemiology of avian influenza A H5N1 virus infection in humans, 1997–2015: A systematic review of individual case data. Lancet Infect. Dis. 2016, 16, e108–e118. [Google Scholar] [CrossRef] [Green Version]
- Goossens, K.E.; Ward, A.C.; Lowenthal, J.W.; Bean, A.G. Chicken interferons, their receptors and interferon-stimulated genes. Dev. Comp. Immunol. 2013, 41, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Downing, T.; Cormican, P.; O‘Farrelly, C.; Bradley, D.G.; Lloyd, A.T. Evidence of the adaptive evolution of immune genes in chicken. BMC Res. Notes 2009, 2, 254. [Google Scholar] [CrossRef] [Green Version]
- Magor, K.E.; Miranzo Navarro, D.; Barber, M.R.; Petkau, K.; Fleming-Canepa, X.; Blyth, G.A.; Blaine, A.H. Defense genes missing from the flight division. Dev. Comp. Immunol. 2013, 41, 377–388. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Huang, B.; Qi, Z.T.; Xu, Z.; Nie, P. Global characterization of interferon regulatory factor (IRF) genes in vertebrates: Glimpse of the diversification in evolution. BMC Immunol. 2010, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Santhakumar, D.; Rubbenstroth, D.; Martinez-Sobrido, L.; Munir, M. Avian Interferons and Their Antiviral Effectors. Front. Immunol. 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Zhu, W.; Ding, C.; Niu, Q.; Wang, H.; Yan, Y.; Sun, J. IRF7 Is Involved in Both STING and MAVS Mediating IFN-beta Signaling in IRF3-Lacking Chickens. J. Immunol. 2019, 203, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Zhou, H. Functional Analysis of Chicken IRF7 in Response to dsRNA Analog Poly(I:C) by Integrating Overexpression and Knockdown. PLoS ONE 2015, 10, e0133450. [Google Scholar] [CrossRef]
- Wang, Z.; Ning, Z.; Sun, M.; Gao, S.; Kang, Y.; Xie, P.; Ren, T. Interferon regulatory factor 7- (IRF7-) mediated immune response affects Newcastle disease virus replication in chicken embryo fibroblasts. Acta Vet. Hung. 2014, 62, 500–511. [Google Scholar] [CrossRef] [Green Version]
- Boutros, M.; Ahringer, J. The art and design of genetic screens: RNA interference. Nat. Rev. Genet. 2008, 9, 554–566. [Google Scholar] [CrossRef]
- Gantz, V.M.; Bier, E. Genome editing. The mutagenic chain reaction: A method for converting heterozygous to homozygous mutations. Science 2015, 348, 442–444. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(∆∆C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. Influenza A virus isolation, culture and identification. Nat. Protoc. 2014, 9, 2663–2681. [Google Scholar] [CrossRef] [Green Version]
- Huprikar, J.; Rabinowitz, S. A simplified plaque assay for influenza viruses in Madin-Darby kidney (MDCK) cells. J. Virol. Methods 1980, 1, 117–120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Webby, R.J.; Woolcock, P.R.; Krauss, S.L.; Webster, R.G. Reassortment and interspecies transmission of North American H6N2 influenza viruses. Virology 2002, 295, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R.; Walther, G.; Hastie, T. Estimating the number of clusters in a data set via the gap statistic. J. Royal Stat. Soc. Ser. B Stat. Methodol. 2001, 63, 411–423. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.; Meir, Y.J.; Coates, C.J.; Handler, A.M.; Pelczar, P.; Moisyadi, S.; Kaminski, J.M. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15008–15013. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Zhou, H. Overexpression of Chicken IRF7 Increased Viral Replication and Programmed Cell Death to the Avian Influenza Virus Infection Through TGF-Beta/FoxO Signaling Axis in DF-1. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Wilk, E.; Pandey, A.K.; Leist, S.R.; Hatesuer, B.; Preusse, M.; Pommerenke, C.; Wang, J.; Schughart, K. RNAseq expression analysis of resistant and susceptible mice after influenza A virus infection identifies novel genes associated with virus replication and important for host resistance to infection. BMC Genom. 2015, 16, 655. [Google Scholar] [CrossRef]
- Ciancanelli, M.J.; Huang, S.X.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Grant, C.E.; Vasa, M.Z.; Deeley, R.G. cIRF-3, a new member of the interferon regulatory factor (IRF) family that is rapidly and transiently induced by dsRNA. Nucleic Acids Res. 1995, 23, 2137–2146. [Google Scholar] [CrossRef] [Green Version]
- Cormican, P.; Lloyd, A.T.; Downing, T.; Connell, S.J.; Bradley, D.; O’Farrelly, C. The avian Toll-Like receptor pathway—Subtle differences amidst general conformity. Dev. Comp. Immunol. 2009, 33, 967–973. [Google Scholar] [CrossRef]
- Crotta, S.; Davidson, S.; Mahlakoiv, T.; Desmet, C.J.; Buckwalter, M.R.; Albert, M.L.; Staeheli, P.; Wack, A. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 2013, 9, e1003773. [Google Scholar] [CrossRef] [Green Version]
- Giotis, E.S.; Ross, C.S.; Robey, R.C.; Nohturfft, A.; Goodbourn, S.; Skinner, M.A. Constitutively elevated levels of SOCS1 suppress innate responses in DF-1 immortalised chicken fibroblast cells. Sci. Rep. 2017, 7, 17485. [Google Scholar] [CrossRef] [Green Version]
- Hatesuer, B.; Hoang, H.T.; Riese, P.; Trittel, S.; Gerhauser, I.; Elbahesh, H.; Geffers, R.; Wilk, E.; Schughart, K. Deletion of Irf3 and Irf7 Genes in Mice Results in Altered Interferon Pathway Activation and Granulocyte-Dominated Inflammatory Responses to Influenza A Infection. J. Innate Immun. 2017, 9, 145–161. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [Green Version]
- Borgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J. Biol. Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Harjunpaa, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Colina, R.; Costa-Mattioli, M.; Dowling, R.J.; Jaramillo, M.; Tai, L.H.; Breitbach, C.J.; Martineau, Y.; Larsson, O.; Rong, L.; Svitkin, Y.V.; et al. Translational control of the innate immune response through IRF-7. Nature 2008, 452, 323–328. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Jones, R.G.; Pearce, E.J. MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells. Immunity 2017, 46, 730–742. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Carlin, A.F.; Plummer, E.M.; Vizcarra, E.A.; Sheets, N.; Joo, Y.; Tang, W.; Day, J.; Greenbaum, J.; Glass, C.K.; Diamond, M.S.; et al. An IRF-3-, IRF-5-, and IRF-7-Independent Pathway of Dengue Viral Resistance Utilizes IRF-1 to Stimulate Type I and II Interferon Responses. Cell Rep. 2017, 21, 1600–1612. [Google Scholar] [CrossRef] [Green Version]
- Karpala, A.J.; Lowenthal, J.W.; Bean, A.G. Activation of the TLR3 pathway regulates IFNβ production in chickens. Dev. Comp. Immunol. 2008, 32, 435–444. [Google Scholar] [CrossRef]
- Karpala, A.J.; Stewart, C.; McKay, J.; Lowenthal, J.W.; Bean, A.G. Characterization of chicken Mda5 activity: Regulation of IFN-β in the absence of RIG-I functionality. J. Immunol. 2011, 186, 5397–5405. [Google Scholar] [CrossRef]
- Cornelissen, J.B.; Post, J.; Peeters, B.; Vervelde, L.; Rebel, J.M. Differential innate responses of chickens and ducks to low-pathogenic avian influenza. Avian Pathol. 2012, 41, 519–529. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, Y.; Lan, X.; Ye, F.; Tian, K.; Zhao, X.; Yin, H.; Li, D.; Xu, H.; Liu, Y.; et al. Molecular characterization, expression of chicken TBK1 gene and its effect on IRF3 signaling pathway. PLoS ONE 2017, 12, e0177608. [Google Scholar] [CrossRef]
- Hiscott, J.; Lin, R.; Nakhaei, P.; Paz, S. MasterCARD: A priceless link to innate immunity. Trends Mol. Med. 2006, 12, 53–56. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.H.; Kern, C.; Zhou, H. Knockout of IRF7 Highlights its Modulator Function of Host Response Against Avian Influenza Virus and the Involvement of MAPK and TOR Signaling Pathways in Chicken. Genes 2020, 11, 385. https://doi.org/10.3390/genes11040385
Kim TH, Kern C, Zhou H. Knockout of IRF7 Highlights its Modulator Function of Host Response Against Avian Influenza Virus and the Involvement of MAPK and TOR Signaling Pathways in Chicken. Genes. 2020; 11(4):385. https://doi.org/10.3390/genes11040385
Chicago/Turabian StyleKim, Tae Hyun, Colin Kern, and Huaijun Zhou. 2020. "Knockout of IRF7 Highlights its Modulator Function of Host Response Against Avian Influenza Virus and the Involvement of MAPK and TOR Signaling Pathways in Chicken" Genes 11, no. 4: 385. https://doi.org/10.3390/genes11040385
APA StyleKim, T. H., Kern, C., & Zhou, H. (2020). Knockout of IRF7 Highlights its Modulator Function of Host Response Against Avian Influenza Virus and the Involvement of MAPK and TOR Signaling Pathways in Chicken. Genes, 11(4), 385. https://doi.org/10.3390/genes11040385