Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
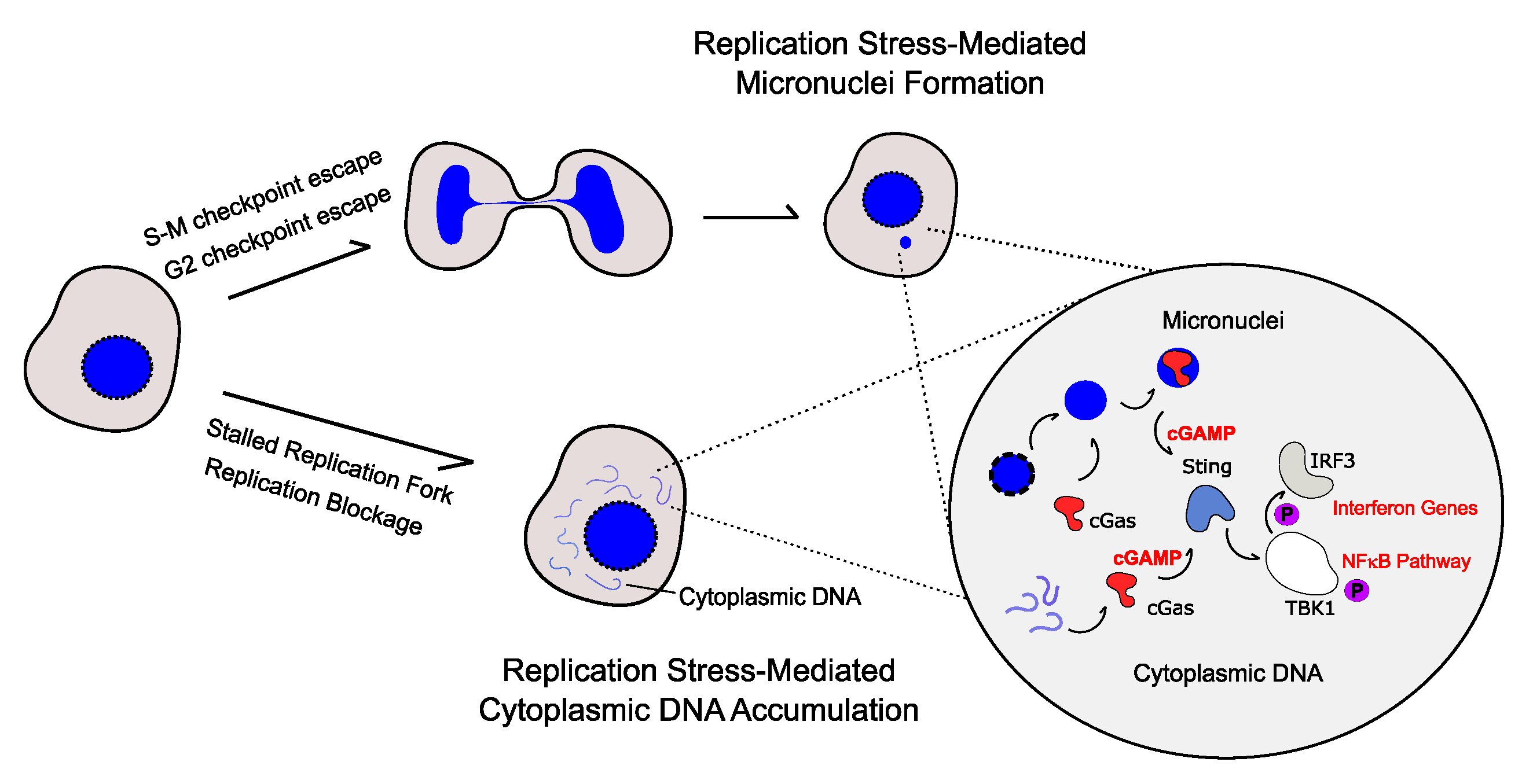
2. Cytoplasmic DNA-Mediated Inflammatory Response
3. Replication Stress Induces the Production of Pro-Inflammatory Cytokines
4. DDR and DNA Repair Defects
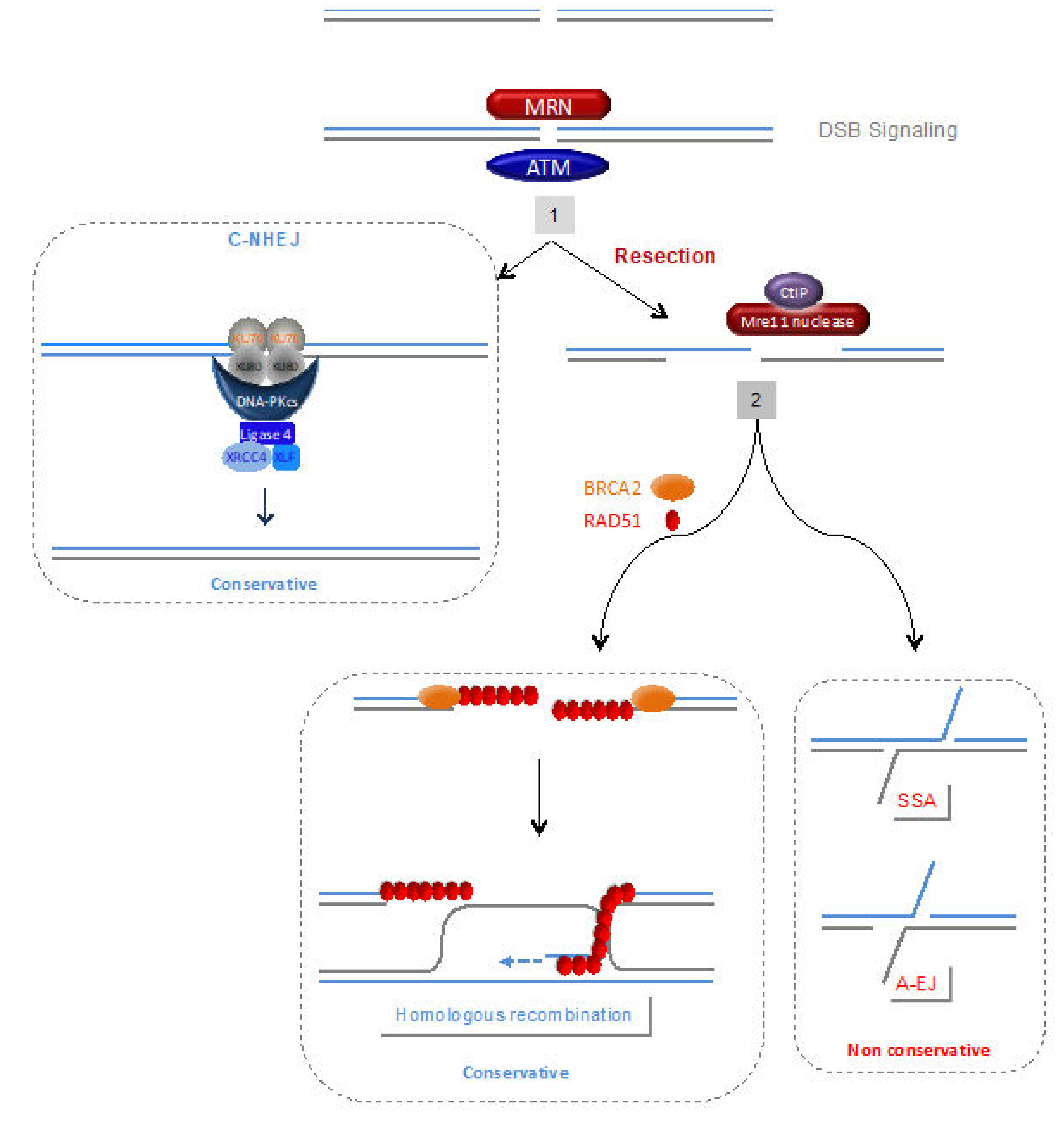
4.1. DNA Damage Signalling and Processing of Double-Strand Ends
4.1.1. Poly-ADP Ribose Polymerase (PARP1)
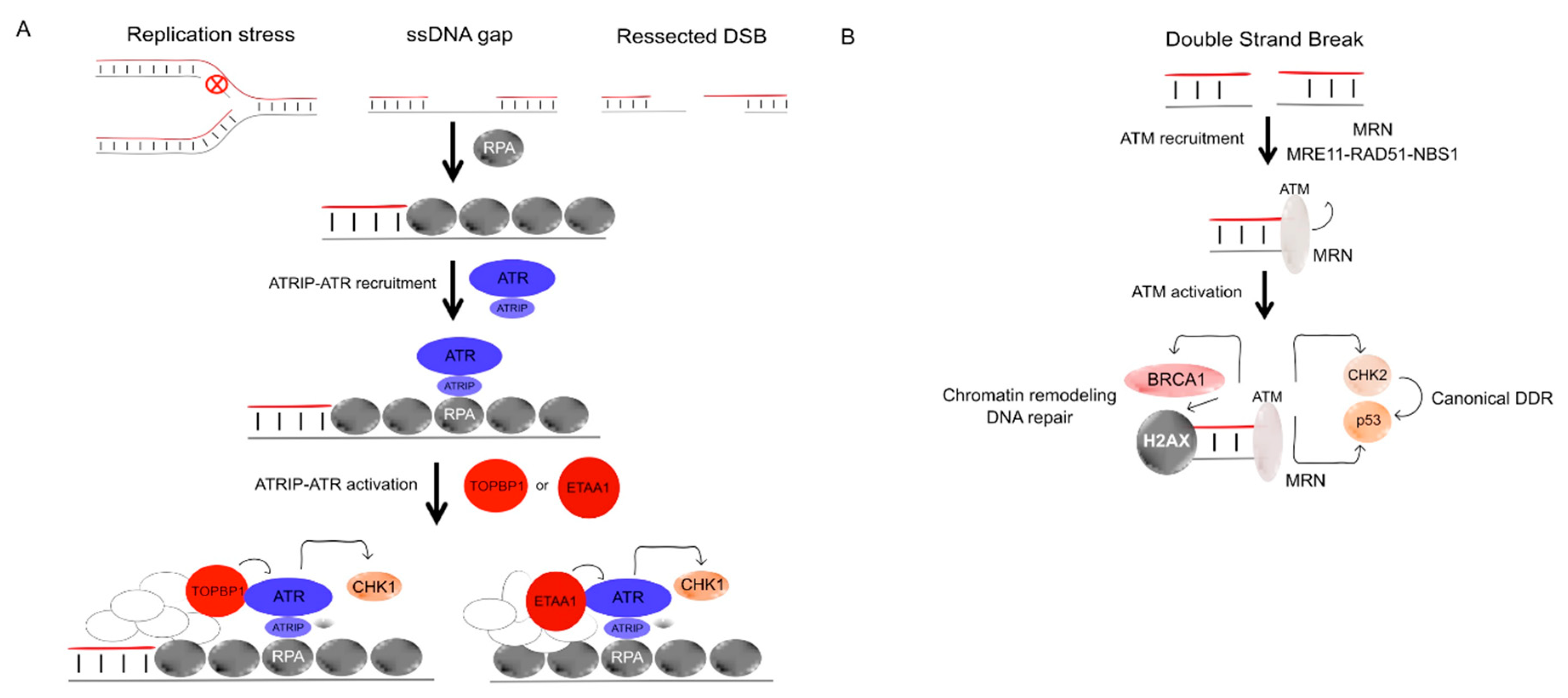
4.1.2. ATR and Chk1
4.1.3. Ataxia Telangiectasia-Mutated (ATM)
4.1.4. The MRN (MRE11/RAD50/NBS1) Complex
4.1.5. SAMHD1 and Aicardi-Goutières Syndrome (AGS)
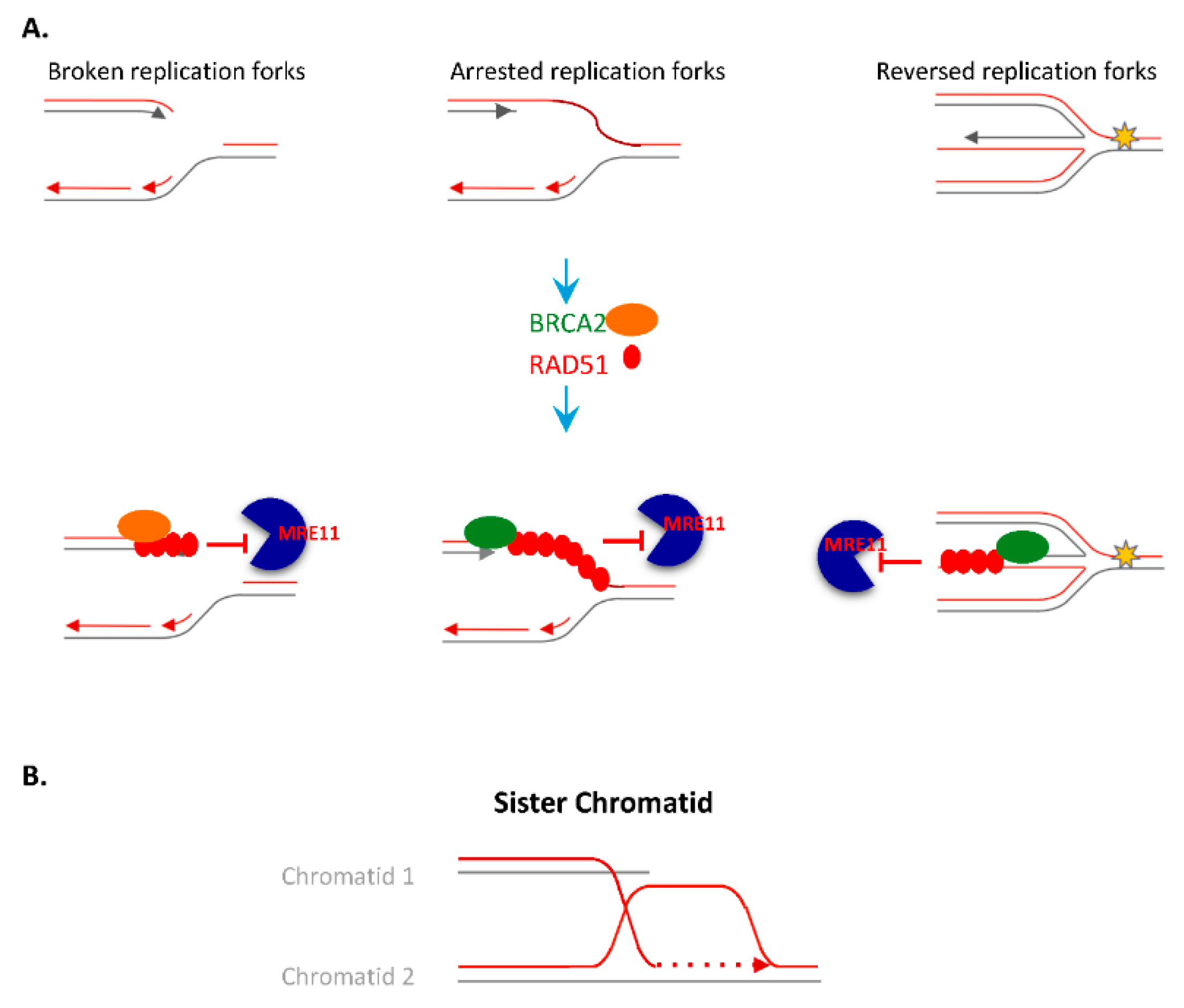
4.2. Homologous Recombination (HR)
4.2.1. Resolution of HR and Arrested Replication Forks Intermediates
a—SLX4 and MUS81
b—RecQ helicases
4.3. C-NHEJ
4.4. Other DNA Repair Pathways
5. DDR and Autoimmune Disease
6. Reactive Oxygen Species and Nitrogen Species
A Vicious Circle
7. Implications for Immunotherapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Técher, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef]
- Somyajit, K.; Gupta, R.; Sedlackova, H.; Neelsen, K.J.; Ochs, F.; Rask, M.B.; Choudhary, C.; Lukas, J. Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science 2017, 358, 797–802. [Google Scholar] [CrossRef]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A.E. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef]
- Dutta, D.; Shatalin, K.; Epshtein, V.; Gottesman, M.E.; Nudler, E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell 2011, 146, 533–543. [Google Scholar] [CrossRef]
- Merrikh, H.; MacHón, C.; Grainger, W.H.; Grossman, A.D.; Soultanas, P. Co-directional replication-transcription conflicts lead to replication restart. Nature 2011, 470, 554–558. [Google Scholar] [CrossRef]
- Sankar, T.S.; Wastuwidyaningtyas, B.D.; Dong, Y.; Lewis, S.A.; Wang, J.D. The nature of mutations induced by replication-transcription collisions. Nature 2016, 535, 178–181. [Google Scholar] [CrossRef]
- Srivatsan, A.; Tehranchi, A.; MacAlpine, D.M.; Wang, J.D. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A Genome-wide siRNA Screen Reveals Diverse Cellular Processes and Pathways that Mediate Genome Stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.J.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Agathangelou, K.; Apostolou, Z.; Garinis, G.A. Nuclear DNA damage and ageing. In Biochemistry and Cell Biology of Ageing: Part I Biomedical Science. Subcellular Biochemistry; Springer: Singapore, 2018; Volume 90, pp. 309–322. [Google Scholar]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2013, 32, 3789–3797. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Wilson, D.M. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, P.F.L.; Schumacher, B. DNA damage responses in ageing. Open Biol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G. Immune Sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Zhang, W.Y.L.; Tan, N.Y.J.; Tripathi, S.; Suter, M.A.; Chew, Z.H.; Khatoo, M.; Ngeow, J.; Cheung, F.S.G. Sensing of dangerous DNA. Mech. Ageing Dev. 2017, 165, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Raulet, D.H. The DNA damage response arouses the immune system. Cancer Res. 2006, 66, 3959–3962. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STI NG pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. CGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363. [Google Scholar] [CrossRef]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. CGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide that Activates Human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Civril, F.; Deimling, T.; De Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Tojima, Y.; Fujimoto, A.; Delhase, M.; Chen, Y.; Hatakeyama, S.; Nakayama, K.; Kaneko, Y.; Nimura, Y.; Motoyama, N.; Ikeda, K.; et al. NAK is an I kappa B kinase-activating kinase. Nature 2000, 404, 778–782. [Google Scholar] [CrossRef]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Hoffmann, E.; Resch, K.; Kracht, M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and c. J. Biol. Chem. 2004, 279, 55633–55643. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-κB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.R.; Bert, N.L.; Ho, S.S.W.; Shen, Y.J.; Tang, M.L.F.; Xiong, G.M.; Croxford, J.L.; Koo, C.X.; Ishii, K.J.; Akira, S.; et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014, 74, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tijhuis, A.E.; Foijer, F. The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells 2019, 8, 1228. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A.; et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Katlinskaya, Y.V.; Carbone, C.J.; Zhao, B.; Katlinski, K.V.; Zheng, H.; Guha, M.; Li, N.; Chen, Q.; Yang, T.; et al. DNA-Damage-Induced Type I Interferon Promotes Senescence and Inhibits Stem Cell Function. Cell Rep. 2015, 11, 785–797. [Google Scholar] [CrossRef]
- Shen, Y.J.; Le Bert, N.; Chitre, A.A.; Koo, C.X.; Nga, X.H.; Ho, S.S.W.; Khatoo, M.; Tan, N.Y.; Ishii, K.J.; Gasser, S. Genome-Derived Cytosolic DNA Mediates Type I Interferon-Dependent Rejection of B Cell Lymphoma Cells. Cell Rep. 2015, 11, 460–473. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Luthra, P.; Aguirre, S.; Yen, B.C.; Pietzsch, C.A.; Sanchez-Aparicio, M.T.; Tigabu, B.; Morlock, L.K.; García-Sastre, A.; Leung, D.W.; Williams, N.S.; et al. Topoisomerase II inhibitors induce DNA damage-dependent interferon responses circumventing ebola virus immune evasion. MBio 2017, 8. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e5. [Google Scholar] [CrossRef]
- Coquel, F.; Neumayer, C.; Lin, Y.L.; Pasero, P. SAMHD1 and the innate immune response to cytosolic DNA during DNA replication. Curr. Opin. Immunol. 2019, 56, 24–30. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [PubMed]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, H.W.; Huttner, D.; Hickson, I.D. How unfinished business from S-phase affects mitosis and beyond. EMBO J. 2013, 32, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Gelot, C.; Magdalou, I.; Lopez, B.S. Replication stress in mammalian cells and its consequences for mitosis. Genes 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Techer, H.; Debatisse, M.; Lopez, B.S. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Bartsch, K.; Knittler, K.; Borowski, C.; Rudnik, S.; Damme, M.; Aden, K.; Spehlmann, M.E.; Frey, N.; Saftig, P.; Chalaris, A.; et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum. Mol. Genet. 2017, 26, 3960–3972. [Google Scholar] [CrossRef]
- MacKenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. CGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Gekara, N.O. DNA damage-induced immune response: Micronuclei provide key platform. J. Cell Biol. 2017, 216, 2999–3001. [Google Scholar] [CrossRef] [PubMed]
- Gratia, M.; Rodero, M.P.; Conrad, C.; Samra, E.B.; Maurin, M.; Rice, G.I.; Duffy, D.; Revy, P.; Petit, F.; Dale, R.C.; et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J. Exp. Med. 2019, 216, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Atianand, M.K.; Fitzgerald, K.A. Molecular Basis of DNA Recognition in the Immune System. J. Immunol. 2013, 190, 1911–1918. [Google Scholar] [CrossRef]
- Wolf, C.; Rapp, A.; Berndt, N.; Staroske, W.; Schuster, M.; Dobrick-Mattheuer, M.; Kretschmer, S.; König, N.; Kurth, T.; Wieczorek, D.; et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Saintigny, Y.; Delacôte, F.; Varès, G.; Petitot, F.; Lambert, S.; Averbeck, D.; Lopez, B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001, 20. [Google Scholar] [CrossRef]
- So, A.; Le Guen, T.; Lopez, B.S.; Guirouilh-Barbat, J. Genomic rearrangements induced by unscheduled DNA double strand breaks in somatic mammalian cells. FEBS J. 2017, 284, 2324–2344. [Google Scholar] [CrossRef]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef]
- Groth, A.; Corpet, A.; Cook, A.J.L.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Pasero, P.; Vindigni, A. Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu. Rev. Genet. 2017, 51, 477–499. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct Mol. Biol 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Huck, S.; Bertrand, P.; Pirzio, L.; Desmaze, C.; Sabatier, L.; Lopez, B.S. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell 2004, 14, 611–623. [Google Scholar] [CrossRef]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10549. [Google Scholar] [CrossRef]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef]
- Dinkelmann, M.; Spehalski, E.; Stoneham, T.; Buis, J.; Wu, Y.; Sekiguchi, J.M.; Ferguson, D.O. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct Mol. Biol 2009, 16, 808–813. [Google Scholar] [CrossRef]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef]
- Le Guen, T.; Ragu, S.; Guirouilh-Barbat, J.; Lopez, B.S. Role of the double-strand break repair pathway in the maintenance of genomic stability. Mol. Cell. Oncol. 2015, 2. [Google Scholar] [CrossRef]
- Haber, J.E. Genome Stability: DNA Repair and Recombination; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Lambert, S.; Lopez, B.S. Characterization of mammalian RAD51 double strand break repair using non lethal dominant negative forms. EMBO J. 2000, 19, 3090–3099. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef]
- Gelot, C.; Guirouilh-Barbat, J.; Lopez, B.S. The Cohesion complex maintains genome stability by preventing the end-joining of distant DNA ends in the S phase. Mol. Cell. Oncol. 2016, 5, e1154123. [Google Scholar] [CrossRef]
- Rooney, S.; Chaudhuri, J.; Alt, F.W. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004, 200, 115–131. [Google Scholar] [CrossRef]
- Stavnezer, J.; Guikema, J.E.J.; Schrader, C.E. Mechanism and Regulation of Class Switch Recombination. Annu. Rev. Immunol. 2008, 26, 261–292. [Google Scholar] [CrossRef]
- Pavri, R. R loops in the regulation of antibody gene diversification. Genes 2017, 8, 154. [Google Scholar] [CrossRef]
- Stavnezer, J.; Schrader, C.E. IgH Chain Class Switch Recombination: Mechanism and Regulation. J. Immunol. 2014, 193, 5370–5378. [Google Scholar] [CrossRef]
- Xu, Z.; Zan, H.; Pone, E.J.; Mai, T.; Casali, P. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat. Rev. Immunol. 2012, 12, 517–531. [Google Scholar] [CrossRef]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Tian, M.; Khuong, C.; Chua, K.; Pinaud, E.; Alt, F.W. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 2003, 422, 726–730. [Google Scholar] [CrossRef]
- Dickerson, S.K.; Market, E.; Besmer, E.; Papavasiliou, F.N. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 2003, 197, 1291–1296. [Google Scholar] [CrossRef]
- Pham, P.; Bransteitter, R.; Petruska, J.; Goodman, M.F. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 2003, 424, 103–107. [Google Scholar] [CrossRef]
- Ramiro, A.R.; Stavropoulos, P.; Jankovic, M.; Nussenzweig, M.C. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 2003, 4, 452–456. [Google Scholar] [CrossRef]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Jeggo, P. Immunological disorders and DNA repair. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2002, 509, 109–126. [Google Scholar] [CrossRef]
- Figueiredo, N.; Chora, A.; Raquel, H.; Pejanovic, N.; Pereira, P.; Hartleben, B.; Neves-Costa, A.; Moita, C.; Pedroso, D.; Pinto, A.; et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity 2013, 39, 874–884. [Google Scholar] [CrossRef]
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair 2014, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Munoz, P.; Malivert, L.; Derdouch, S.; Azerrad, C.; Abramowski, V.; Revy, P.; de Villartay, J.P. DNA repair and the immune system: From V(D)J recombination to aging lymphocytes. Eur. J. Immunol. 2007, 37. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Martin-Hernandez, K.; Rodriguez-Vargas, J.M.; Schreiber, V.; Dantzer, F. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin. Cell Dev. Biol. 2017, 63, 92–101. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.A.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination–mediated DNA repair. Cancer Discov. 2014, 4, 1430–1447. [Google Scholar] [CrossRef]
- Hochegger, H.; Dejsuphong, D.; Fukushima, T.; Morrison, C.; Sonoda, E.; Schreiber, V.; Guang, Y.Z.; Saberi, A.; Masutani, M.; Adachi, N.; et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006, 25, 1305–1314. [Google Scholar] [CrossRef]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, L.; Barbeau, J.; Curtin, N.J.; Morris, E.P.; Pearl, L.H. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012, 40, 4168–4177. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef]
- Liu, Y.; Kadyrov, F.A.; Modrich, P. PARP-1 enhances the mismatch-dependence of 5’-directed excision in human mismatch repair in vitro. DNA Repair 2011, 10, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, F.; De La Rubia, G.; Ménissier-De Murcia, J.; Hostomsky, Z.; De Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking poly(ADP- ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef]
- Prasad, R.; Lavrik, O.I.; Kim, S.J.; Kedar, P.; Yang, X.P.; Vande Berg, B.J.; Wilson, S.H. DNA polymerase β-mediated long patch base excision repair: Poly(ADP-ribose) polymerase-1 stimulates strand displacement DNA synthesis. J. Biol. Chem. 2001, 276, 32411–32414. [Google Scholar] [CrossRef]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The Enzymatic and DNA Binding Activity of PARP-1 Are Not Required for NF-κB Coactivator Function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-κB in inflammatory disorders. Cell. Mol. Life Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef]
- Oliver, F.J.; Ménissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; De La Rubia, G.; Stoclet, J.C.; De Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef] [PubMed]
- Mangerich, A.; Bürkle, A. Pleiotropic cellular functions of PARP1 in longevity and aging: Genome maintenance meets inflammation. Oxid. Med. Cell. Longev. 2012, 2012, 321653. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Haenni, S.S.; Buerki, C.; Meier, N.I.; Lane, W.S.; Owen, H.; Gersbach, M.; Imhof, R.; Hottiger, M.O. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-κB-dependent transcription. J. Biol. Chem. 2005, 280, 40450–40464. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Brégnard, C.; Guerra, J.; Déjardin, S.; Passalacqua, F.; Benkirane, M.; Laguette, N. Upregulated LINE-1 Activity in the Fanconi Anemia Cancer Susceptibility Syndrome Leads to Spontaneous Pro-inflammatory Cytokine Production. EBioMedicine 2016, 8, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, T.; Murano, H.; Nakamura, M.; Ichinohe, K.; Tanaka, K. Activation of interferon-stimulated genes by g-Ray irradiation independently of the ataxia telangiectasia mutated-p53 pathway. Mol. Cancer Res. 2011, 9, 476–484. [Google Scholar] [CrossRef]
- Eickmeier, O.; Kim, S.Y.; Herrmann, E.; Döring, C.; Duecker, R.; Voss, S.; Wehner, S.; Hölscher, C.; Pietzner, J.; Zielen, S.; et al. Altered mucosal immune response after acute lung injury in a murine model of Ataxia Telangiectasia. BMC Pulm. Med. 2014, 14. [Google Scholar] [CrossRef]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell 2018, 71, 419–427. [Google Scholar] [CrossRef]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef]
- Takeda, S.; Nakamura, K.; Taniguchi, Y.; Paull, T.T. Ctp1/CtIP and the MRN Complex Collaborate in the Initial Steps of Homologous Recombination. Mol. Cell 2007, 28, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA Repair 2010, 9, 1283–1291. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Deshpande, R.A. The Mre11/Rad50/Nbs1 complex: Recent insights into catalytic activities and ATP-driven conformational changes. Exp. Cell Res. 2014, 329, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Lopes, S.; Tur, J.; Calatayud-Subias, J.A.; Lloberas, J.; Stracker, T.H.; Celada, A. NBS1 is required for macrophage homeostasis and functional activity in mice. Blood 2015, 126, 2502–2510. [Google Scholar] [CrossRef]
- Prochazkova, J.; Sakaguchi, S.; Owusu, M.; Mazouzi, A.; Wiedner, M.; Velimezi, G.; Moder, M.; Turchinovich, G.; Hladik, A.; Gurnhofer, E.; et al. DNA Repair Cofactors ATMIN and NBS1 Are Required to Suppress T Cell Activation. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef]
- Seidel, P.; Remus, M.; Delacher, M.; Grigaravicius, P.; Reuss, D.E.; Frappart, L.; von Deimling, A.; Feueuer, M.; Abdollahi, A.; Frappart, P.O. Epidermal Nbn deletion causes premature hair loss and a phenotype resembling psoriasiform dermatitis. Oncotarget 2016, 7, 23006–23018. [Google Scholar] [CrossRef][Green Version]
- Roth, S.; Rottach, A.; Lotz-Havla, A.S.; Laux, V.; Muschaweckh, A.; Gersting, S.W.; Muntau, A.C.; Hopfner, K.P.; Jin, L.; Vanness, K.; et al. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat. Immunol. 2014, 15, 538–545. [Google Scholar] [CrossRef]
- Kondo, T.; Kobayashi, J.; Saitoh, T.; Maruyama, K.; Ishii, K.J.; Barber, G.N.; Komatsu, K.; Akira, S.; Kawai, T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type i interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 2013, 110, 2969–2974. [Google Scholar] [CrossRef]
- Rice, G.I.; Rodero, M.P.; Crow, Y.J. Human Disease Phenotypes Associated With Mutations in TREX1. J. Clin. Immunol. 2015, 35, 235–243. [Google Scholar] [CrossRef]
- Crow, Y.J.; Rehwinkel, J. Aicardi-Goutie’res syndrome and related phenotypes: Linking nucleic acid metabolism with autoimmunity. Hum. Mol. Genet. 2009, 18. [Google Scholar] [CrossRef] [PubMed]
- Orcesi, S.; La Piana, R.; Fazzi, E. Aicardi-Goutires syndrome. Br. Med. Bull. 2009, 89, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Chahwan, C.; Chahwan, R. Aicardi-Goutieres syndrome: From patients to genes and beyond. Clin. Genet. 2012, 81, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gall, A.; Treuting, P.; Elkon, K.B.; Loo, Y.M.; Gale, M.; Barber, G.N.; Stetson, D.B. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity 2012, 36, 120–131. [Google Scholar] [CrossRef]
- Ahn, J.; Barber, G.N. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr. Opin. Immunol. 2014, 31, 121–126. [Google Scholar] [CrossRef]
- Gray, E.E.; Treuting, P.M.; Woodward, J.J.; Stetson, D.B. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi–Goutières Syndrome. J. Immunol. 2015, 195, 1939–1943. [Google Scholar] [CrossRef]
- Gao, D.; Li, T.; Li, X.D.; Chen, X.; Li, Q.Z.; Wight-Carter, M.; Chen, Z.J. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA 2015, 112, E5699–E5705. [Google Scholar] [CrossRef]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; Van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef]
- Pokatayev, V.; Hasin, N.; Chon, H.; Cerritelli, S.M.; Sakhuja, K.; Ward, J.M.; Douglas Morris, H.; Yan, N.; Crouch, R.J. RNase H2 catalytic core Aicardi-Goutières syndrome-Related mutant invokes cGAS-STING innate immunesensing pathway in mice. J. Exp. Med. 2016, 213, 329–336. [Google Scholar] [CrossRef]
- Daddacha, W.; Koyen, A.E.; Bastien, A.J.; Head, P.S.E.; Dhere, V.R.; Nabeta, G.N.; Connolly, E.C.; Werner, E.; Madden, M.Z.; Daly, M.B.; et al. SAMHD1 Promotes DNA End Resection to Facilitate DNA Repair by Homologous Recombination. Cell Rep. 2017, 20, 1921–1935. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Olopade, O.I. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer 2007, 7, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian ovarian cancer study group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Daboussi, F.; Courbet, S.; Benhamou, S.; Kannouche, P.; Zdzienicka, M.Z.; Debatisse, M.; Lopez, B.S. A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 2008, 121, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-dependent innate immune signaling by s-phase-specific DNA damage in breast cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-β Responses. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef]
- Gangloff, S.; Soustelle, C.; Fabre, F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 2000, 25, 192–194. [Google Scholar] [CrossRef]
- Ciccia, A.; McDonald, N.; West, S.C. Structural and Functional Relationships of the XPF/MUS81 Family of Proteins. Annu. Rev. Biochem. 2008, 77, 259–287. [Google Scholar] [CrossRef]
- Ho, S.S.W.; Zhang, W.Y.L.; Tan, N.Y.J.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.G.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef]
- Dufour, C.; Corcione, A.; Svahn, J.; Haupt, R.; Poggi, V.; Béka’ssy, A.N.; Scimè, R.; Pistorio, A.; Pistoia, V. TNF-α and IFN-γ are overexpressed in the bone marrow of Fanconi anemia patients and TNF-α suppresses erythropoiesis in vitro. Blood 2003, 102, 2053–2059. [Google Scholar] [CrossRef]
- Sumpter, R.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s syndrome: Clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Grabarz, A.; Guirouilh-Barbat, J.J.; Barascu, A.A.; Pennarun, G.G.; Genet, D.; Rass, E.; Germann, S.M.; Bertrand, P.; Hickson, I.D.; Lopez, B.S. A role for BLM in double-strand break repair pathway choice: Prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol. Biomarkers Prev. 1996, 5, 239–246. [Google Scholar]
- Goto, M. Hierarchical deterioration of body systems in Werner’s syndrome: Implications for normal ageing. Mech. Ageing Dev. 1997, 98, 239–254. [Google Scholar] [CrossRef]
- Goto, M. Werner’s syndrome: From clinics to genetics. Clin. Exp. Rheumatol. 2000, 18, 760–766. [Google Scholar] [PubMed]
- Turaga, R.V.N.; Paquet, E.R.; Sild, M.; Vignard, J.; Garand, C.; Johnson, F.B.; Masson, J.Y.; Lebel, M. The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell Cycle 2009, 8, 2080–2092. [Google Scholar] [CrossRef]
- Lebel, M.; Massip, L.; Garand, C.; Thorin, E. Ascorbate improves metabolic abnormalities in Wrn mutant mice but not the free radical scavenger catechin. Ann. N Y Acad. Sci. 2010, 1197, 40–44. [Google Scholar]
- Goto, M.; Sugimoto, K.; Hayashi, S.; Ogino, T.; Sugimoto, M.; Furuichi, Y.; Matsuura, M.; Ishikawa, Y.; Iwaki-Egawa, S.; Watanabe, Y. Aging-associated inflammation in healthy Japanese individuals and patients with Werner syndrome. Exp. Gerontol. 2012, 47, 936–939. [Google Scholar] [CrossRef]
- Van der Burg, M.; IJspeert, H.; Verkaik, N.; Turul, T.; Wiegant, W.; Morotomi-Yano, K.; Mari, P.-O.; Tezcan, I.; Chen, D.; Zdzienicka, M.; et al. A DNA-PKcs mutation in a radiosensitive T–B– SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Invest. 2008, 119, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.K.; Alt, F.W.; Yeap, L.-S. Related Mechanisms of Antibody Somatic Hypermutation and Class Switch Recombination. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.-W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting Edge: Ku70 Is a Novel Cytosolic DNA Sensor That Induces Type III Rather Than Type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef]
- Nakad, R.; Schumacher, B. DNA damage response and immune defense: Links and mechanisms. Front. Genet. 2016, 7. [Google Scholar] [CrossRef]
- Diderich, K.; Alanazi, M.; Hoeijmakers, J.H.J. Premature aging and cancer in nucleotide excision repair-disorders. DNA Repair 2011, 10, 772–780. [Google Scholar] [CrossRef]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci. 2009, 66, 994–1009. [Google Scholar] [CrossRef]
- De Boer, J.; Hoeijmakers, J.H.J. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef]
- David, S.S.; Williams, S.D. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998, 98, 1221–1261. [Google Scholar] [CrossRef]
- Huffman, J.L.; Sundheim, O.; Tainer, J.A. DNA base damage recognition and removal: New twists and grooves. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 577, 55–76. [Google Scholar] [CrossRef]
- Dalhus, B.; Laerdahl, J.K.; Backe, P.H.; Bjørås, M. DNA base repair—Recognition and initiation of catalysis. FEMS Microbiol. Rev. 2009, 33, 1044–1078. [Google Scholar] [CrossRef] [PubMed]
- Hang, B. Base excision repair. In DNA Repair, Genetic Instability, and Cancer; World Scientific: Singapore, 2007; pp. 23–64. ISBN 9789812706782. [Google Scholar]
- Fromme, J.C.; Verdine, G.L. Base excision repair. Adv. Protein Chem. 2004, 69, 1–41. [Google Scholar] [PubMed]
- Peña-Diaz, J.; Jiricny, J. Mammalian mismatch repair: Error-free or error-prone? Trends Biochem. Sci. 2012, 37, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Karakasilioti, I.; Kamileri, I.; Chatzinikolaou, G.; Kosteas, T.; Vergadi, E.; Robinson, A.R.; Tsamardinos, I.; Rozgaja, T.A.; Siakouli, S.; Tsatsanis, C.; et al. DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab. 2013, 18, 403–415. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. NF-κB inhibition delays DNA damage—Induced senescence and aging in mice. J. Clin. Invest. 2012, 122, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, T.A.; Fontes, F.L.; Coutinho, L.G.; de Souza, F.R.S.; de Melo, J.T.A.; de Souto, J.T.; Leib, S.L.; Agnez-Lima, L.F. SNPs in DNA repair genes associated to meningitis and host immune response. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 713, 39–47. [Google Scholar] [CrossRef]
- Touati, E.; Michel, V.; Thiberge, J.M.; Avé, P.; Huerre, M.; Bourgade, F.; Klungland, A.; Labigne, A. Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 2006, 11, 494–505. [Google Scholar] [CrossRef]
- Guikema, J.E.J.; Linehan, E.K.; Esa, N.; Tsuchimoto, D.; Nakabeppu, Y.; Woodland, R.T.; Schrader, C.E. AP endonuclease 2 regulates the expansion of germinal centers by protecting against AID-independent DNA damage in B cells 1. J. Immunol. 2014, 15, 931–939. [Google Scholar] [CrossRef]
- Fung, H.; Demple, B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell 2005, 17, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 1–5. [Google Scholar] [CrossRef]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef]
- Cavazzana, I.; Ceribelli, A.; Quinzanini, M.; Scarsi, M.; Airò, P.; Cattaneo, R.; Franceschini, F. Prevalence and clinical associations of anti-Ku antibodies in systemic autoimmune diseases. Lupus 2008, 17, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Schild-Poulter, C.; Su, A.; Shih, A.; Kelly, O.P.; Fritzler, M.J.; Goldstein, R.; Haché, R.J.G. Association of autoantibodies with Ku and DNA repair proteins in connective tissue diseases. Rheumatology 2008, 47, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.C.; Pettijohn, K.; Fike, F.; Wang, J.; Nahas, S.A.; Tunuguntla, R.; Hu, H.; Gatti, R.A.; McCurdy, D. Defective DNA double-strand break repair in pediatric systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Bawadekar, M.; De Andrea, M.; Lo Cigno, I.; Baldanzi, G.; Caneparo, V.; Graziani, A.; Landolfo, S.; Gariglio, M. The Extracellular IFI16 Protein Propagates Inflammation in Endothelial Cells Via p38 MAPK and NF-κB p65 Activation. J. Interf. Cytokine Res. 2015, 35, 441–453. [Google Scholar] [CrossRef]
- Commoner, B.; Townsend, J.; Pake, G.E. Free radicals in biological materials. Nature 1954, 174, 689–691. [Google Scholar] [CrossRef]
- Wallace, S.S. Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med. 2002, 33, 1–14. [Google Scholar] [CrossRef]
- Kawanishi, S.; Hiraku, Y. Oxidative and nitrative DNA damage as biomarker for carcinogenesis with special reference to inflammation. Antioxid. Redox Signal. 2006, 8, 1047–1058. [Google Scholar] [CrossRef]
- Murata, M.; Thanan, R.; Ma, N.; Kawanishi, S. Role of nitrative and oxidative DNA damage in inflammation-related carcinogenesis. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Oikawa, S.; Murata, M. Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells. Genes Environ. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Ameziane-El-Hassani, R.; Schlumberger, M.; Dupuy, C. NADPH oxidases: New actors in thyroid cancer? Nat. Rev. Endocrinol. 2016, 12, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A. Respiratory burst in human neutrophils. J. Immunol. Methods 1999, 232, 3–14. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Perry, J.J.P.; Shin, D.S.; Getzoff, E.D.; Tainer, J.A. The structural biochemistry of the superoxide dismutases. Biochim. Biophys. Acta Proteins Proteomics 2010, 1804, 245–262. [Google Scholar] [CrossRef]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)-induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011, 711, 167–173. [Google Scholar] [CrossRef]
- Won, J.K.; Bakhoum, S.F. The cytosolic DNA-sensing cGAS–sting pathway in cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tüting, T.; Hartmann, G.; Barchet, W. Oxidative damage of dna confers resistance to cytosolic nuclease trex1 degradation and potentiates STING-dependent immune sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, oxidative stress and innate immunity. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Gerber, S.; Lord, E. The role of Type I interferons in antitumor responses: Implications in radiation therapy. J. Immunol. 2011, 186. [Google Scholar]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xiao, X.; Yang, X.; Zhang, Z.; Wu, L.; Liu, Z. STING signaling in tumorigenesis and cancer therapy: A friend or foe? Cancer Lett. 2017, 402, 203–212. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef]
- Ahn, J.; Xia, T.; Konno, H.; Konno, K.; Ruiz, P.; Barber, G.N. Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Man, S.M.; Gurung, P.; Liu, Z.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.-D. Cutting Edge: STING Mediates Protection against Colorectal Tumorigenesis by Governing the Magnitude of Intestinal Inflammation. J. Immunol. 2014, 193, 4779–4782. [Google Scholar] [CrossRef] [PubMed]
- Gaston, J.; Cheradame, L.; Yvonnet, V.; Deas, O.; Poupon, M.F.; Judde, J.G.; Cairo, S.; Goffin, V. Intracellular STING inactivation sensitizes breast cancer cells to genotoxic agents. Oncotarget 2016, 7, 77205–77224. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; van Vugt, M.A.T.M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell–intrinsic immunity in ERCC1-deficient non–small cell lung cancer. J. Clin. Invest. 2019, 129, 1211–1228. [Google Scholar] [CrossRef]
- Ubhi, T.; Brown, G.W. Exploiting DNA replication stress for cancer treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409. https://doi.org/10.3390/genes11040409
Ragu S, Matos-Rodrigues G, Lopez BS. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes. 2020; 11(4):409. https://doi.org/10.3390/genes11040409
Chicago/Turabian StyleRagu, Sandrine, Gabriel Matos-Rodrigues, and Bernard S. Lopez. 2020. "Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response" Genes 11, no. 4: 409. https://doi.org/10.3390/genes11040409
APA StyleRagu, S., Matos-Rodrigues, G., & Lopez, B. S. (2020). Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes, 11(4), 409. https://doi.org/10.3390/genes11040409