Gallop Racing Shifts Mature mRNA towards Introns: Does Exercise-Induced Stress Enhance Genome Plasticity?

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Training
2.2. Sampling
2.3. RNA Extraction
2.4. Sequencing
2.5. Bioinformatic Analyses
2.5.1. Annotations Retrieval and Count Matrices
2.5.2. Differential Expression Analyses: Genes
2.5.3. Differential Expression Analyses: Isoforms
2.5.4. Differential Expression Analyses: Repetitive Elements
- (1)
- Genome-wide differential expression analysis
- (2)
- Differential expression analysis of repeats classes
- (3)
- Differential expression analysis of long interspersed nuclear elements subclass 1 (LINE1) only.
3. Results
3.1. Sequencing Statistics
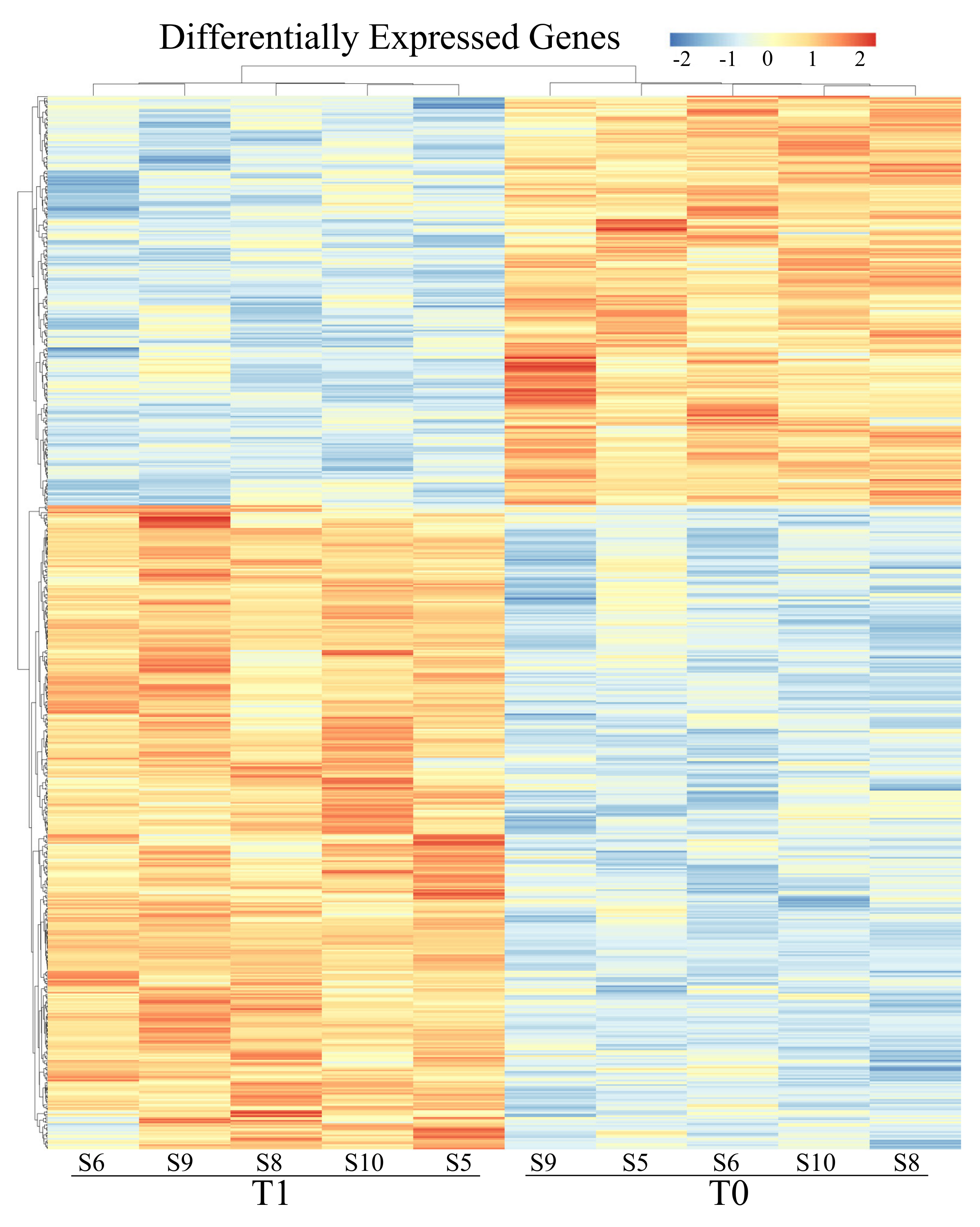
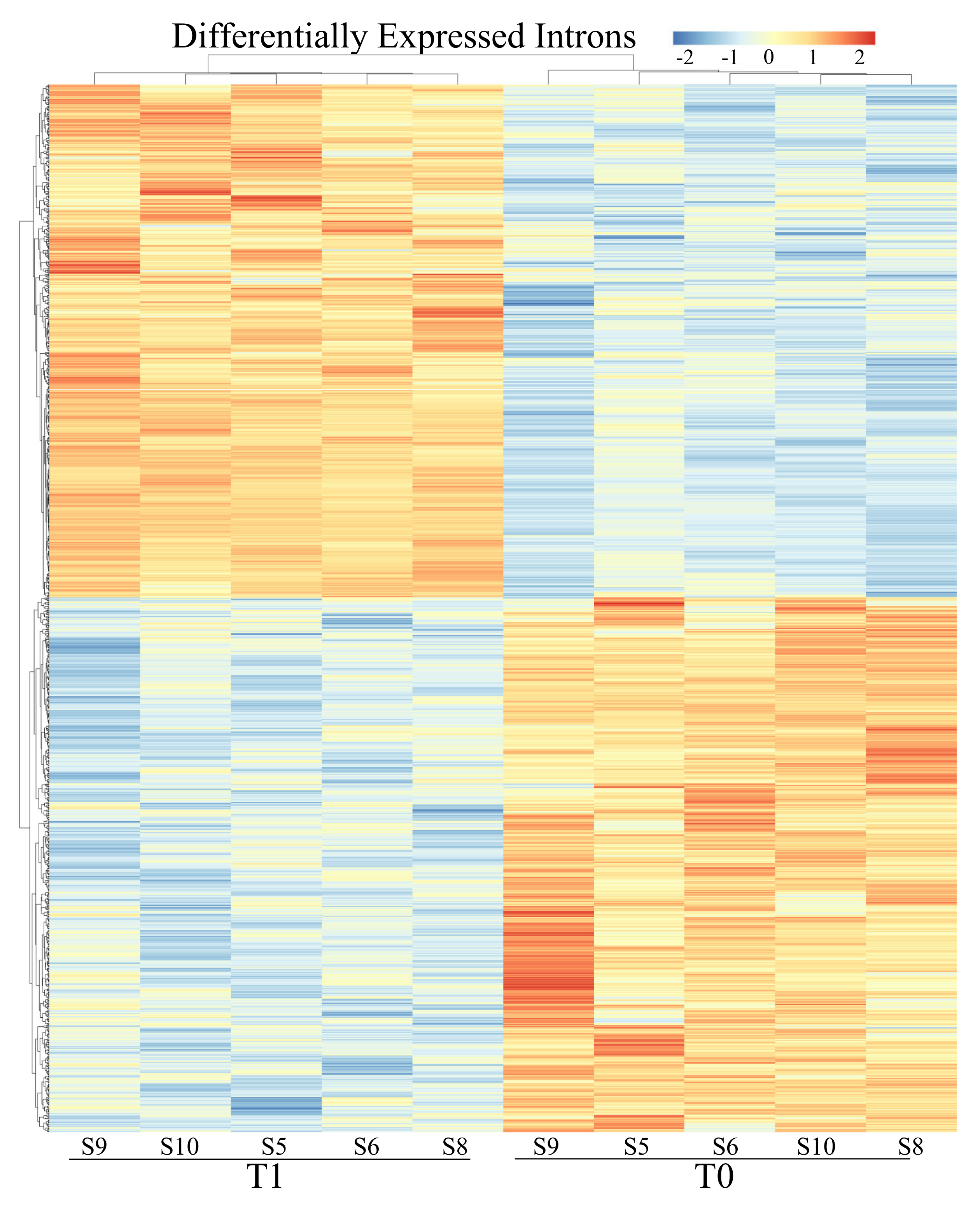
3.2. Differential Expression Analyses: Genes
3.3. Differential Expression Analyses: Isoforms
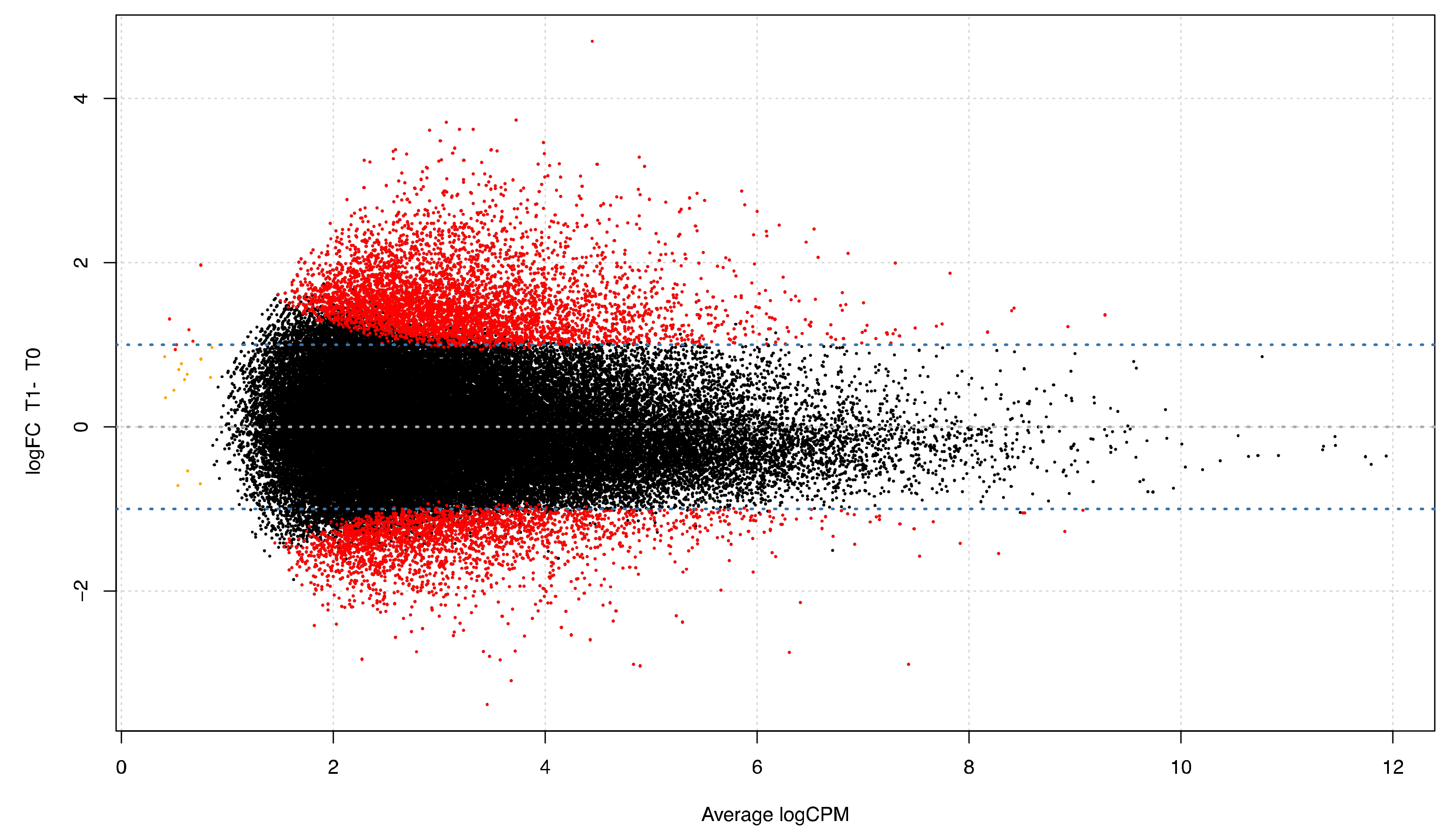
3.4. Differential Expression Analyses: Repetitive Elements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Giuffra, E.; Tuggle, C.K. and Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 2019, 7, 65–88. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.; Shin, J.W.; Carninci, P. Paradigm shifts in genomics through the FANTOM projects. Mamm. Genome 2015, 26, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, H.; Foreman, J. Heart and spleen weights as a function of breed and somatotype. Equine Exerc Physiol. 1991, 3, 17–21. [Google Scholar]
- Barrey, E.; Galloux, P.; Valette, J.P.; Auvinet, B.; Wolter, R. Stride Characteristics of Overground versus Treadmill Locomotion in the Saddle Horse. Cells Tissues Organs 1993, 146, 90–94. [Google Scholar] [CrossRef]
- Hargreaves, B.J.; Kronfeld, D.S.; Naylor, J.R.J. Ambient temperature and relative humidity influenced packed cell volume, total plasma protein and other variables in horses during an incremental submaximal field exercise test. Equine Vet. J. 1999, 31, 314–318. [Google Scholar] [CrossRef]
- Cappelli, K.; Felicetti, M.; Capomaccio, S.; Nocelli, C.; Silvestrelli, M.; Verini-Supplizi, A. Effect of training status on immune defence related gene expression in Thoroughbred: Are genes ready for the sprint? Vet. J. 2013, 195, 373–376. [Google Scholar] [CrossRef]
- Ohmura, H.; Mukai, K.; Takahashi, T.; Aida, H.; Jones, J.H. Cardiorespiratory function in Thoroughbreds during locomotion on a treadmill at an incline or decline. Am. J. Vet. Res. 2017, 78, 340–349. [Google Scholar] [CrossRef]
- Rivero, J.L.L.; van Breda, E.; Rogers, C.W.; Lindner, A.; Sloet van Oldruitenborgh-Oosterbaan, M.M. Unexplained underperformance syndrome in sport horses: Classification, potential causes and recognition. Equine Vet. J. 2008, 40, 611–618. [Google Scholar] [CrossRef]
- Cappelli, K.; Supplizi, A.V.; Capomaccio, S.; Albertini, E.; Silvestrelli, M. Analysis Of Peripheral Blood Mononuclear Cells Gene Expression in Endurance Horses; Town & Country Convention Center: San Diego, CA, USA, 2005. [Google Scholar]
- Cappelli, K.; Verini-Supplizi, A.; Capomaccio, S.; Silvestrelli, M. Analysis of peripheral blood mononuclear cells gene expression in endurance horses by cDNA-AFLP technique. Res. Vet. Sci. 2007, 82, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, K.; Felicetti, M.; Capomaccio, S.; Spinsanti, G.; Silvestrelli, M.; Verini Supplizi, A. Exercise induced stress in horses: Selection of the most stable reference genes for quantitative RT-PCR normalization. BMC Mol. Biol. 2008, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelli, K.; Felicetti, M.; Capomaccio, S.; Pieramati, C.; Silvestrelli, M.; Verini-Supplizi, A. Exercise-induced up-regulation of MMP-1 and IL-8 genes in endurance horses. BMC Physiol. 2009, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capomaccio, S.; Cappelli, K.; Barrey, E.; Felicetti, M.; Silvestrelli, M.; Verini-Supplizi, A. Microarray analysis after strenuous exercise in peripheral blood mononuclear cells of endurance horses. Anim. Genet. 2010, 41, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Capomaccio, S.; Cappelli, K.; Spinsanti, G.; Mencarelli, M.; Muscettola, M.; Felicetti, M.; Supplizi, A.; Bonifazi, M. Athletic humans and horses: Comparative analysis of interleukin-6 (IL-6) and IL-6 receptor (IL-6R) expression in peripheral blood mononuclear cells in trained and untrained subjects at rest. BMC Physiol. 2011, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Capomaccio, S.; Vitulo, N.; Verini-Supplizi, A.; Barcaccia, G.; Albiero, A.; D’Angelo, M.; Campagna, D.; Valle, G.; Felicetti, M.; Silvestrelli, M.; et al. RNA Sequencing of the Exercise Transcriptome in Equine Athletes. PLoS ONE 2013, 8, e83504. [Google Scholar]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Braunschweig, U.; Barbosa-Morais, N.L.; Pan, Q.; Nachman, E.N.; Alipanahi, B.; Gonatopoulos-Pournatzis, T.; Frey, B.; Irimia, M.; Blencowe, B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014, 24, 1774–1786. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as regulators of gene expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [Green Version]
- Capomaccio, S.; Verini-Supplizi, A.; Galla, G.; Vitulo, N.; Barcaccia, G.; Felicetti, M.; Silvestrelli, M.; Cappelli, K. Transcription of LINE-derived sequences in exercise-induced stress in horses. Anim. Genet. 2010, 41, 23–27. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O′Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Saremi, N.F.; et al. Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biol. 2018, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Brief. Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Vilborg, A.; Steitz, J.A. Readthrough transcription: How are DoGs made and what do they do? RNA Biol. 2016, 14, 632–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecinka, A.; Mittelsten Scheid, O. Stress-Induced Chromatin Changes: A Critical View on Their Heritability. Plant Cell Physiol. 2012, 53, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biamonti, G.; Caceres, J.F. Cellular stress and RNA splicing. Trends Biochem. Sci. 2009, 34, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Kaer, K.; Branovets, J.; Hallikma, A.; Nigumann, P.; Speek, M. Intronic L1 Retrotransposons and Nested Genes Cause Transcriptional Interference by Inducing Intron Retention, Exonization and Cryptic Polyadenylation. PLoS ONE 2011, 6, e26099. [Google Scholar] [CrossRef]
- Sznajder, Ł.J.; Thomas, J.D.; Carrell, E.M.; Reid, T.; McFarland, K.N.; Cleary, J.D.; Oliveira, R.; Nutter, C.A.; Bhatt, K.; Sobczak, K.; et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. USA 2018, 115, 4234–4239. [Google Scholar] [CrossRef] [Green Version]
- Muotri, A.R.; Marchetto, M.C.N.; Coufal, N.G.; Gage, F.H. The necessary junk: New functions for transposable elements. Hum. Mol. Genet. 2007, 16, R159–R167. [Google Scholar] [CrossRef] [Green Version]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Herzel, L.; Neugebauer, K.M. Quantification of co-transcriptional splicing from RNA-Seq data. Methods 2015, 85, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J. Alternative Splicing: New Insights from Global Analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ZHENG, C.L.; FU, X.-D.; GRIBSKOV, M. Characteristics and regulatory elements defining constitutive splicing and different modes of alternative splicing in human and mouse. RNA 2005, 11, 1777–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, K.; Green, P. Differing patterns of selection in alternative and constitutive splice sites. Genome Res. 2007, 17, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Shalgi, R.; Hurt, J.A.; Lindquist, S.; Burge, C.B. Widespread Inhibition of Posttranscriptional Splicing Shapes the Cellular Transcriptome following Heat Shock. Cell Rep. 2014, 7, 1362–1370. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, K.; Adachi, S.; Natsume, T.; Iwakiri, J.; Terai, G.; Asai, K.; Hirose, T. LncRNA-dependent nuclear stress bodies promote intron retention through SR protein phosphorylation. EMBO J. 2019, 39, e102729. [Google Scholar]
- Anufrieva, K.S.; Shender, V.О.; Arapidi, G.P.; Pavlyukov, M.S.; Shakhparonov, M.I.; Shnaider, P.V.; Butenko, I.O.; Lagarkova, M.A.; Govorun, V.M. Therapy-induced stress response is associated with downregulation of pre-mRNA splicing in cancer cells. Genome Med. 2018, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Brady, L.K.; Wang, H.; Radens, C.M.; Bi, Y.; Radovich, M.; Maity, A.; Ivan, C.; Ivan, M.; Barash, Y.; Koumenis, C. Transcriptome analysis of hypoxic cancer cells uncovers intron retention in EIF2B5 as a mechanism to inhibit translation. PLoS Biol. 2017, 15, e2002623. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [CrossRef] [Green Version]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome Sequence, Comparative Analysis, and Population Genetics of the Domestic Horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crichton, J.H.; Dunican, D.S.; MacLennan, M.; Meehan, R.R.; Adams, I.R. Defending the genome from the enemy within: Mechanisms of retrotransposon suppression in the mouse germline. Cell. Mol. Life Sci. 2014, 71, 1581–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.R.; Doucet, A.J.; Kopera, H.C.; Moldovan, J.B.; Garcia-Pérez, J.L.; Moran, J.V. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol. Spectr. 2015, 3, 1165–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aporntewan, C.; Phokaew, C.; Piriyapongsa, J.; Ngamphiw, C.; Ittiwut, C.; Tongsima, S.; Mutirangura, A. Hypomethylation of Intragenic LINE-1 Represses Transcription in Cancer Cells through AGO2. PLoS ONE 2011, 6, e17934. [Google Scholar] [CrossRef]
- Wongpaiboonwattana, W.; Tosukhowong, P.; Dissayabutra, T.; Mutirangura, A.; Boonla, C. Oxidative Stress Induces Hypomethylation of LINE-1 and Hypermethylation of the RUNX3 Promoter in a Bladder Cancer Cell Line. Asian Pac. J. Cancer Prev. 2013, 14, 3773–3778. [Google Scholar] [CrossRef] [Green Version]
- Miousse, I.R.; Koturbash, I. The Fine LINE: Methylation Drawing the Cancer Landscape. Available online: https://www.hindawi.com/journals/bmri/2015/131547/ (accessed on 5 November 2019).
- Scott, E.; Devine, S. The Role of Somatic L1 Retrotransposition in Human Cancers. Viruses 2017, 9, 131. [Google Scholar] [CrossRef]
- Kroutter, E.N.; Belancio, V.P.; Wagstaff, B.J.; Roy-Engel, A.M. The RNA Polymerase Dictates ORF1 Requirement and Timing of LINE and SINE Retrotransposition. PLoS Genet 2009, 5, e1000458. [Google Scholar] [CrossRef] [Green Version]
- Ostertag, E.M.; Kazazian, H.H., Jr. Biology of Mammalian L1 Retrotransposons. Annu. Rev. Genet. 2001, 35, 501–538. [Google Scholar] [CrossRef]
- Pizarro, J.G.; Cristofari, G. Post-Transcriptional Control of LINE-1 Retrotransposition by Cellular Host Factors in Somatic Cells. Front. Cell Dev. Biol. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Mackinnon, L.T. Chronic exercise training effects on immune function. Med. Sci. Sports Exerc. 2000, 32, S369. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Reads before Trimming | Reads after Trimming | Quality Check Passed Rate (%) | Uniquely Mapped Reads | Alignment Rate (%) |
|---|---|---|---|---|---|
| S5_T0 | 18,986,960 | 18,391,294 | 96.9 | 7,930,946 | 86.2 |
| S5_T1 | 19,209,286 | 18,644,550 | 97.1 | 8,112,987 | 87.0 |
| S6_T0 | 19,143,702 | 18,542,082 | 96.9 | 7,833,923 | 84.4 |
| S6_T1 | 15,681,066 | 15,223,494 | 97.1 | 6,619,839 | 87.0 |
| S8_T0 | 20,252,456 | 19,654,560 | 97.0 | 8,542,631 | 86.9 |
| S8_T1 | 31,767,674 | 30,848,206 | 97.1 | 13,349,560 | 86.6 |
| S9_T0 | 15,724,850 | 15,241,064 | 96.9 | 6,598,277 | 86.6 |
| S9_T1 | 25,053,756 | 24,340,766 | 97.2 | 10,589,195 | 86.7 |
| S10_T0 | 20,330,330 | 19,746,518 | 97.1 | 8,631,228 | 87.4 |
| S10_T1 | 33,503,070 | 32,465,956 | 96.9 | 14,217,209 | 87.4 |
| Average | 21,965,315 | 21,309,849 | 97.0 | 9,242,580 | 86.60 |
| Sample | Total Alignments | Successfully Assigned Alignments EXONS | % | Successfully Assigned Alignments INTRONS | % | Successfully Assigned Alignments REPEATS | % |
|---|---|---|---|---|---|---|---|
| S10_T0 | 9,418,426 | 5,220,406 | 55.4 | 2,282,112 | 24.2 | 1,397,171 | 14.8 |
| S5_T0 | 8,731,754 | 4,697,224 | 53.8 | 2,233,626 | 25.6 | 1,342,363 | 15.4 |
| S6_T0 | 8,600,396 | 4,899,846 | 57 | 1,863,096 | 21.7 | 1,145,141 | 13.3 |
| S8_T0 | 9,313,192 | 5,146,610 | 55.3 | 2,268,959 | 24.4 | 1,423,386 | 15.3 |
| S9_T0 | 7,236,941 | 4,332,077 | 59.9 | 1,442,060 | 19.9 | 921,749 | 12.7 |
| Average | 8,660,141.80 | 4,859,232.60 | 56.28 | 2,017,970.60 | 23.16 | 1,245,962.00 | 14.30 |
| S10_T1 | 15,479,241 | 8,015,665 | 51.8 | 4,616,059 | 29.8 | 2,603,246 | 16.8 |
| S5_T1 | 8,858,376 | 4,813,955 | 54.3 | 2,389,440 | 27 | 1,377,956 | 15.6 |
| S6_T1 | 7,230,807 | 3,883,746 | 53.7 | 1,976,927 | 27.3 | 1,138,745 | 15.7 |
| S8_T1 | 14,650,606 | 7,835,416 | 53.5 | 3,893,566 | 26.6 | 2,256,959 | 15.4 |
| S9_T1 | 11,642,249 | 6,259,752 | 53.8 | 3,167,804 | 27.2 | 1,806,643 | 15.5 |
| Average | 11,572,255.80 | 6,161,706.80 | 53.42 | 3,208,759.20 | 27.58 | 1,836,709.80 | 15.80 |
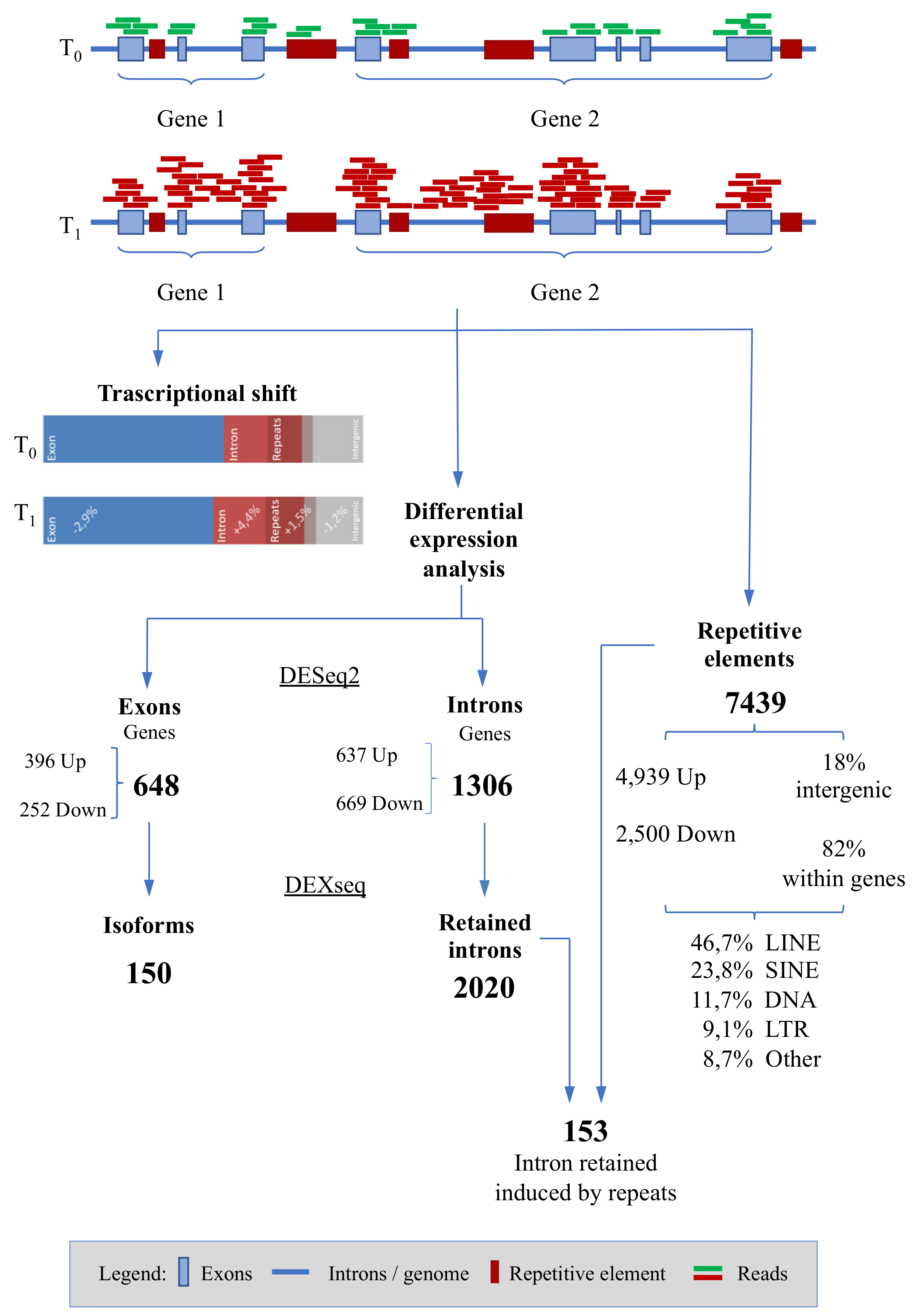
| Race VS. Basal | −2.86 | +4.42 | +1.50 | ||||
| t-Test | 0.029 | 0.008 | 0.028 |
| Genes | Introns | |||
|---|---|---|---|---|
| ID | log2Fold Change | ID | log2Fold Change | |
| Upregulated | ENSECAG00000030595 | 10.53 | ENSECAG00000018841 | 6.29 |
| ENSECAG00000019352 | 7.58 | ENSECAG00000039315 | 6.09 | |
| ENSECAG00000001516 | 6.98 | ENSECAG00000020003 | 5.39 | |
| ENSECAG00000038063 | 6.24 | ENSECAG00000017073 | 4.79 | |
| ENSECAG00000023163 | 6.24 | ENSECAG00000011929 | 4.49 | |
| ENSECAG00000009129 | 5.97 | ENSECAG00000004515 | 4.33 | |
| ENSECAG00000009755 | 5.96 | ENSECAG00000002619 | 4.30 | |
| ENSECAG00000021383 | 5.75 | ENSECAG00000010669 | 4.00 | |
| ENSECAG00000011929 | 5.57 | ENSECAG00000005905 | 3.95 | |
| ENSECAG00000020402 | 5.54 | ENSECAG00000030110 | 3.91 | |
| ENSECAG00000034297 | 5.48 | ENSECAG00000003573 | 3.86 | |
| ENSECAG00000039315 | 5.45 | ENSECAG00000010860 | 3.77 | |
| ENSECAG00000033016 | 5.34 | ENSECAG00000016321 | 3.68 | |
| ENSECAG00000015766 | 4.67 | ENSECAG00000013594 | 3.66 | |
| ENSECAG00000020003 | 4.53 | ENSECAG00000000051 | 3.66 | |
| ENSECAG00000040244 | 4.51 | ENSECAG00000039959 | 3.57 | |
| ENSECAG00000002234 | 4.36 | ENSECAG00000014979 | 3.56 | |
| ENSECAG00000010860 | 4.31 | ENSECAG00000023173 | 3.47 | |
| ENSECAG00000033856 | 4.31 | ENSECAG00000009215 | 3.43 | |
| ENSECAG00000015992 | 4.00 | ENSECAG00000015318 | 3.37 | |
| Downregulated | ENSECAG00000032106 | −23.04 | ENSECAG00000040402 | −4.91 |
| ENSECAG00000034632 | −6.08 | ENSECAG00000023475 | −4.33 | |
| ENSECAG00000007460 | −5.64 | ENSECAG00000028489 | −4.14 | |
| ENSECAG00000021087 | −4.55 | ENSECAG00000011895 | −3.99 | |
| ENSECAG00000010281 | −4.17 | ENSECAG00000031371 | −3.62 | |
| ENSECAG00000009895 | −4.07 | ENSECAG00000032756 | −3.58 | |
| ENSECAG00000035315 | −3.54 | ENSECAG00000036032 | −3.56 | |
| ENSECAG00000008274 | −3.40 | ENSECAG00000036704 | −3.48 | |
| ENSECAG00000009869 | −3.22 | ENSECAG00000000386 | −3.45 | |
| ENSECAG00000032544 | −3.09 | ENSECAG00000017676 | −3.43 | |
| ENSECAG00000032503 | −3.04 | ENSECAG00000025038 | −3.34 | |
| ENSECAG00000030934 | −2.90 | ENSECAG00000022510 | −3.34 | |
| ENSECAG00000034974 | −2.85 | ENSECAG00000006114 | −3.28 | |
| ENSECAG00000009625 | −2.78 | ENSECAG00000027794 | −3.26 | |
| ENSECAG00000034925 | −2.72 | ENSECAG00000011660 | −3.18 | |
| ENSECAG00000010326 | −2.68 | ENSECAG00000014338 | −3.12 | |
| ENSECAG00000036608 | −2.63 | ENSECAG00000033795 | −3.10 | |
| ENSECAG00000037661 | −2.55 | ENSECAG00000033519 | −2.99 | |
| ENSECAG00000014338 | −2.53 | ENSECAG00000011723 | −2.97 | |
| ENSECAG00000015083 | −2.48 | ENSECAG00000036290 | −2.85 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappelli, K.; Mecocci, S.; Gioiosa, S.; Giontella, A.; Silvestrelli, M.; Cherchi, R.; Valentini, A.; Chillemi, G.; Capomaccio, S. Gallop Racing Shifts Mature mRNA towards Introns: Does Exercise-Induced Stress Enhance Genome Plasticity? Genes 2020, 11, 410. https://doi.org/10.3390/genes11040410
Cappelli K, Mecocci S, Gioiosa S, Giontella A, Silvestrelli M, Cherchi R, Valentini A, Chillemi G, Capomaccio S. Gallop Racing Shifts Mature mRNA towards Introns: Does Exercise-Induced Stress Enhance Genome Plasticity? Genes. 2020; 11(4):410. https://doi.org/10.3390/genes11040410
Chicago/Turabian StyleCappelli, Katia, Samanta Mecocci, Silvia Gioiosa, Andrea Giontella, Maurizio Silvestrelli, Raffaele Cherchi, Alessio Valentini, Giovanni Chillemi, and Stefano Capomaccio. 2020. "Gallop Racing Shifts Mature mRNA towards Introns: Does Exercise-Induced Stress Enhance Genome Plasticity?" Genes 11, no. 4: 410. https://doi.org/10.3390/genes11040410
APA StyleCappelli, K., Mecocci, S., Gioiosa, S., Giontella, A., Silvestrelli, M., Cherchi, R., Valentini, A., Chillemi, G., & Capomaccio, S. (2020). Gallop Racing Shifts Mature mRNA towards Introns: Does Exercise-Induced Stress Enhance Genome Plasticity? Genes, 11(4), 410. https://doi.org/10.3390/genes11040410