3.2. The Transcriptome of gpx5 Mutants Under Laboratory Growth Conditions
To assess the transcriptome of the
gpx5 mutant under optimized laboratory growth conditions (no extra oxidative stress added), we performed RNA-seq on CC4348 and
gpx5 cells grown at a density of 2 × 10
6 cells mL
−1 in exponential phase in TAP complete medium at 25 °C under constant illumination of 60 µmol·m
−2∙s
−1 with continuous shaking at 120 rpm. The experimental design and analyses are shown in
Figure S2. The sequence reads were aligned, and expression level were determined based on the
Chlamydomonas reference genome [
30,
31] (
Table S1). Differentially expressed genes (DEGs) were screened with the criteria of false discovery rate (FDR) ≤ 0.05, fold change ≥ 2, and at least half of transcript abundance from pairwise comparisons ≥ 10 reads per kb of exon per million fragments (RPKM). According to these criteria, we identified 880 DEGs in the
gpx5 mutant compared to the parental strain CC4348 under optimized growth conditions, of which 380 DEGs were upregulated and 500 were downregulated (
Table S2).
These DEGs were classified by using MapMan [
32], employing the Algal Functional Annotation Tool [
33] (
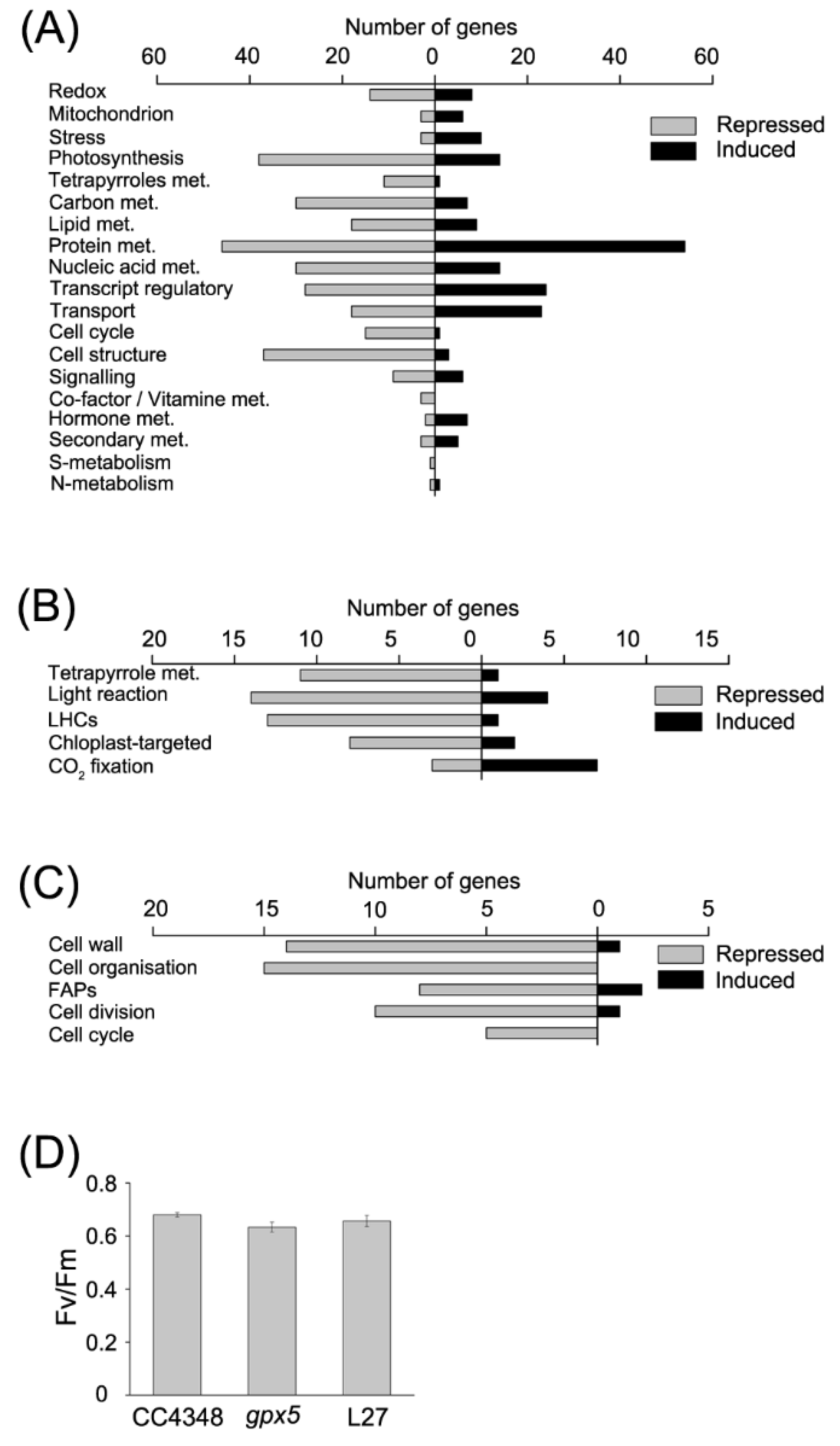
Figure 2A). We first analyzed transcripts involved in redox metabolism, among which the abundance of 8 transcripts increased, and the abundance of 14 transcripts decreased in the
gpx5 mutant compared to CC4348. The
GPX5 transcript was hardly detectable, as expected.
GPX4, another member of the GPX family, had a slightly higher transcript abundance in the
gpx5 mutant than in CC4348. The other members of GPX family,
GPX1–GPX3, had no significant change in transcript abundance in the
gpx5 mutant. These data suggested that the expression of other GPX proteins were not upregulated to compensate for the lack of GPX5.
Among the eight upregulated transcripts related to redox in the
gpx5 mutant under optimized growth conditions, the transcripts of two nucleoredoxins (
NRX2 and
NRX3) and one sulfiredoxin (
SRX1) increased by more than 4.5-fold. As antioxidants, NRXs reduce other proteins by cysteine thiol-disulfide exchange, functioning as protein disulfide reductases (PDIs). Similarly,
PDI5 transcript abundance significantly increased more than two-fold in the
gpx5 mutant. NRX2, NRX3, SRX1, and PDI5 can reduce the disulfide bond of oxidized proteins, attenuating the effects of oxidative stress. The abundance of transcripts of
VTC2 (Cre13.g588150), encoding GDP-L-galactose phosphorylase, a key enzyme in de novo ascorbate synthesis [
34], increased by about three-fold in the
gpx5 mutant, suggesting that the synthesis of the antioxidant ascorbate (ASC) was induced. These data suggested that some mechanisms of ROS scavenging were enhanced in the
gpx5 mutant, and that, even under unstressed mixotrophic conditions, the presence of ROS occurred and was sufficient for cell signaling.
In order to detect the effects of GPX5 deficiency on basal metabolism, we found the modifications in the CCM occurred in the mutant. Most of genes encoding proteins involving in CCM were upregulated in the gpx5 mutant, including some low-CO2-inducible proteins (CCP2, LCI24, LCI5, LCIB, and LCIE). Moreover, we also analyzed the expression change of genes in the chlororespiraton. Among them, seven out of nine transcripts showed no difference in abundance between CC4348 and the gpx5 mutant, including glycolate dehydrogenase (GYD1, Cre06.g288700), phosphoglycolate phosphatase (PGP1 and PGP2 encoding by Cre03.g168700 and Cre10.g438100, respectively), serine hydroxymethyltransferase (SHMT1, Cre16.g664550), alanine-glyoxylate transaminase (AGT1 and AGT2), and serine glyoxylate aminotransferase (SGA1). Only hydroxypyruvate reductase (HPR1) and alanine aminotransferase (AAT1) were downregulated in the gpx5 mutant. These results meant the deficiency of GPX5 had few effects on chlororespiraton.
Transcript abundances of the glutathione
S-transferase (GST) genes (
GST1 and
GST10) were very low in the
gpx5 mutant (4 and 6 RPKM, respectively) compared to that of either GST1 or GST10 in CC4348. GSTs detoxify endogenous or xenobiotic compounds and prevent oxidative damage to cells [
35,
36,
37]. Stress-inducible GSTs with glutathione peroxidase activity can protect plants from oxidative injury [
38]. Moreover, GSTS are biomarkers of the response to stress and overexpression of GSTs enhances tolerance to abiotic stress [
39,
40]. The transcript abundance of
GSTs in the
gpx5 mutant was not higher than that in CC4348. This suggests that the
gpx5 mutant was not experiencing oxidative stress when grown on TAP complete medium under our optimized laboratory growth conditions.
Mutation of
GPX5 resulted in the adjustment of metabolic processes. The transcripts of 38 photosynthesis-related genes were decreased and 14 photosynthesis-related transcripts were increased in the
gpx5 mutant. Most of the transcripts encoding antenna proteins showed a downward trend, except the
LHCA3 transcript, encoding a chlorophyll a/b binding protein of light-harvesting complex (LHC) I, which increased about 2-fold in abundance in the
gpx5 mutant. Moreover, nearly all of the genes involved in tetrapyrrole synthesis were downregulated, except for
UPS1, which encodes uroporphyrinogen-III synthase (Cre09.g409100) (
Figure 2A,B). To confirm the effect of GPX5 deficiency on photosynthesis, we measured the maximum efficiency of photosystem II (PSII) (
Figure 2D). The results showed a slightly reduction in photosynthetic efficiency of the
gpx5 mutant compared to CC4348, suggesting that the
gpx5 mutant might minimize ROS accumulation by slowing down photosynthesis, which is a major source of ROS.
Among 37 DEGs encoding proteins related to carbon metabolism, including glycolysis, gluconeogenesis, starch synthesis, starch degradation, and the TCA cycle, 30 transcripts were downregulated in the gpx5 mutant. The transcript with the largest decrease in level encodes malate synthase (MAS1), which uses acetyl coenzyme A to synthesize malic acid. The abundance of the MAS1 transcript decreased from 756 RPKM in CC4348 to 221 RPKM in the gpx5 mutant. We also detected the isocitrate lyase (ICL1) transcript, which decreased in abundance from 785RPKM in CC4348 to 282 RPKM in the gpx5 mutant under laboratory growth conditions; and so did the transcripts of citrate synthase (CIS2) and acetyl-CoA syntheses/ligase (ACS3). The transcript of CIS2 and ACS3 decreased from 80 to 62 RPKM, and from 2508 to 1361 RPKM, respectively. These data would simply suggest that, since gpx5 is growing more slowly than the control, the transcripts encoding transcripts involved in the glyoxylate cycle are in lower amounts. Almost all genes encoding enzymes in the glycolysis/gluconeogenesis pathway were downregulated in the gpx5 mutant, except fructose-1,6-bisphosphate aldolase (FBA1). Nevertheless, the reaction from fructose-1,6-diphosphate to glyceraldehyde-3-phosphate catalyzed by FBA1 is reversible, so it might not affect the overall metabolic slowdown of the glycolysis/gluconeogenesis pathway.
Lack of GPX5 also affected lipid metabolism, resulting in 27 lipid-related DEGs (9 upregulated and 18 downregulated). The transcript abundance of
KAS2 and
KAS3, which encode β ketoacyl acyl-carrier-protein synthase II and synthase III, respectively, and
HAD1, encoding 3-hydroxyacyl-carrier-protein dehydratase, decreased to 0.07- to 0.35-fold in the
gpx5 mutant. The transcripts of
FAD6, encoding chloroplast omega-6-fatty acid desaturase, increased in abundance by 2.3-fold, reaching 1374 RPKM. Increasing the unsaturation of membrane lipids in cyanobacteria can improve the tolerance to low temperature [
41]. Therefore, upregulation of the expression of genes encoding fatty acid desaturases may affect the metabolism of unsaturated fatty acid in the
gpx5 mutant.
Functional classification showed that 15 out of 18 DEGs related to amino acid metabolism were downregulated in the gpx5 mutant. The abundance of transcripts encoding the enzymes involved in arginine synthesis were significantly decreased, including arginosuccinate synthase (Cre09.g416050, AGS1), acetylglutamate kinase (Cre01.g015000, AGK1), argininosuccinate lyase (Cre01.g021251, ARG7), N-acetylglutamate synthase (Cre16.g694850, NGS1), and N-acetyl-gamma-glutamyl-phosphate reductase (Cre03.g146187). These observations suggested that decreasing synthesis of arginine might play some function in response to GPX5 deficiency or the resulting ROS imbalance.
Besides the effect of oxidative stress on metabolism, transcripts related to the cell cycle and structural proteins were significantly downregulated in the
gpx5 mutant (
Figure 2A,C). These effect on cell-cycle-related genes is consistent with the effect on growth described above (
Figure 1C). In our previous study [
15], we found that the
gpx5 mutation died earlier than the parental stain CC4348 when cultured in nitrogen-free medium. The grow difference between the mutant
gpx5 and parental strain suggested that the deficiency of GPX5 in the mutant not only affected ROS homeostasis, but also affected cell growth.
We also identified DEGs related to nucleotide or nucleic acid metabolism, transcript regulatory, mitochondrial electron transport chain (mETC) or encoding transporters. For example, the transcripts encoding a bZIP transcription factor BLZ8 (Cre12.g501600) and a zinc finger transcription factor (Cre03.g156250) increased about 2.5-fold, with transcript abundance increasing from 108 and 105 RPKM in CC4348 to 258 and 253 RPKM in the gpx5 mutant, respectively.
In summary, intracellular antioxidant metabolism and transport appeared to adjust to maintain redox homeostasis in the gpx5 mutant. The expression of genes related to carbon metabolism, lipid metabolism, and synthesis of chlorophyll were depressed, and the efficiency of photosynthesis decreased in the gpx5 mutant rather than in CC4348. These overall multifaceted changes in transcription slowed the growth of the gpx5 mutant cells.
3.3. Transcriptome Responses to 1O2 in Parental Strain CC4348 Cells
To address the role of GPX5 in response to oxidative stress, we first studied the mechanisms by which parental strain
C. reinhardtii cells respond to oxidative stress. To this end, we performed RNA-seq on CC4348 cells at 0, 30, and 60 min after the addition of 1 μM RB to the medium. Using the same criteria as before, we identified 708 DEGs after the 30-min RB treatment, accounting for 4.7% of the total genome. These DEGs included 322 upregulated transcripts and 386 downregulated transcripts (
Table S3).
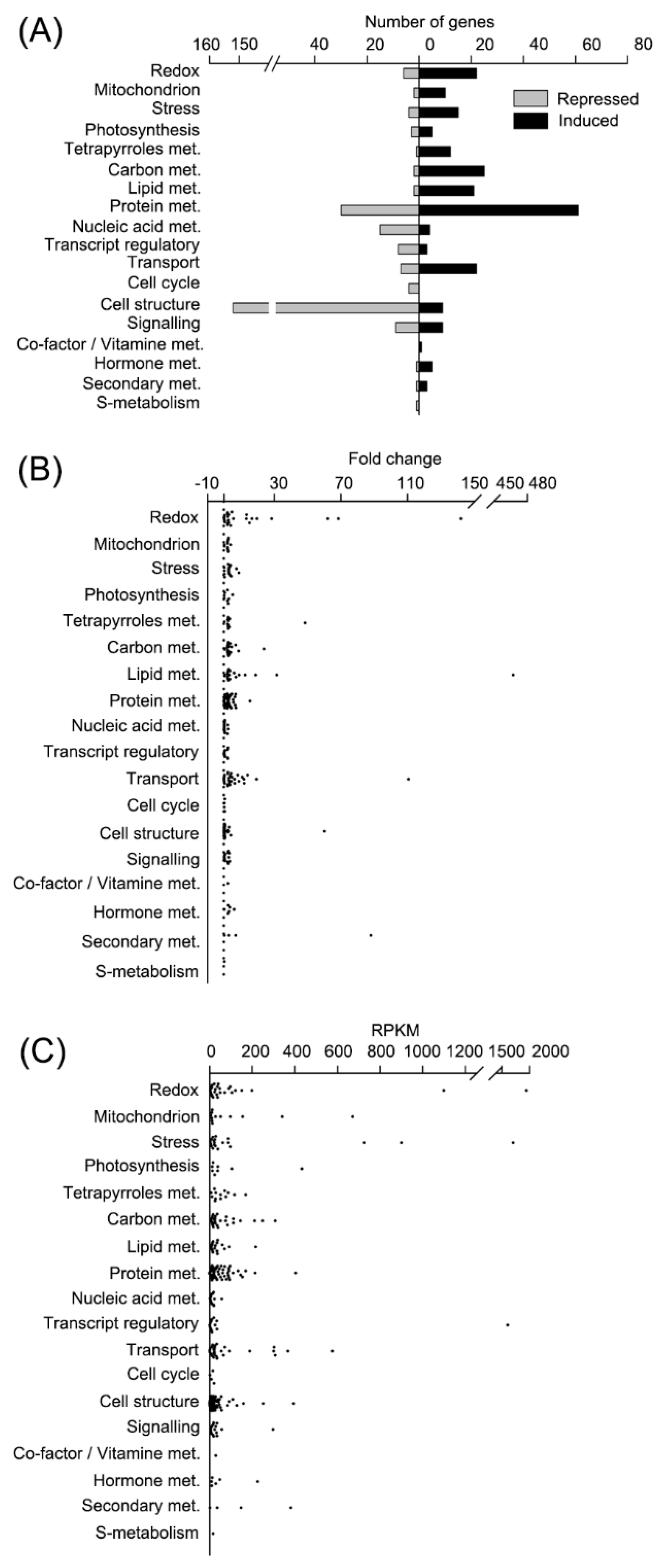
ROS homeostasis was disturbed in CC4348 cells under oxidative stress. The transcripts of 22 redox metabolism-related genes were upregulated, and six were downregulated (
Figure 3A). The transcript abundances of genes encoding ROS-scavenging enzymes, such as ascorbate peroxidase (APX1), glutathione reductase (GSR1), GPX5, and the GST family, were upregulated after
1O
2 stress.
GPX5 has been reported to be highly induced by
1O
2 [
11,
12], and the transcript abundance of
GPX5 increased 13.5-fold after treatment with RB for 30 min in CC4348 cells (
Table S3). Another non-selenocysteine GPX gene (
GPX4) and selenium-dependent GPX gene (
GPX2) showed no difference in transcript abundance. Additionally, the transcript of
GPX3 was barely detectable. Interestingly, one selenium-dependent GPX gene (
GPX1) showed the opposite expression pattern, as it was downregulated from 100 to 6 RPKM after 30 min of RB treatment. The active site of GPX1 is selenocysteine, which is easily oxidized and has strong reducibility. Generally, antioxidants function actively during oxidative stress, so the downregulation of
GPX1 was unexpected. One possible explanation is that different
GPX genes are induced by different oxidative stresses.
We noted that most genes involved in redox metabolism were upregulated in CC4348 cells after treatment with 1 μM RB for 30 min. Among the 28 redox-related DEGs, the transcripts of
ferritin2, encoding a ferritin-like protein (Cre01.g033300) had the greatest fold change, from 14 to 1944 RPKM, an increase of about 140-fold (
Figure 3B and
Table S3). Ranked second were SOUL heme-binding protein genes (
SOUL1 and
SOUL2), whose transcripts increased more than 60-fold. This indicated that heme might play an important role in maintaining redox homeostasis. Moreover,
GST family genes also responded to RB treatment, including Cre07.g342100,
GST1 (Cre16.g688550),
GST10 (Cre12.g559800), and
PDO2 (Cre12.g559800) which belong to the GST KAPPA class. Among them, Cre07.g342100 encodes a GST, whose transcript increased sharply by 29-fold within 30 min after
1O
2 stress, reaching a peak of 121 RPKM. Thioredoxin superfamily proteins and proteins containing thioredoxin domains, including NRX2, protein disulfide-isomerase A6 (PDIA6, Cre07.g326600), and r53.5-related protein LCI7 (Cre06.g263550) also responded to
1O
2. Among them,
PDIA6 and
LCI7 were upregulated more than 15-fold within 30 min of the treatment.
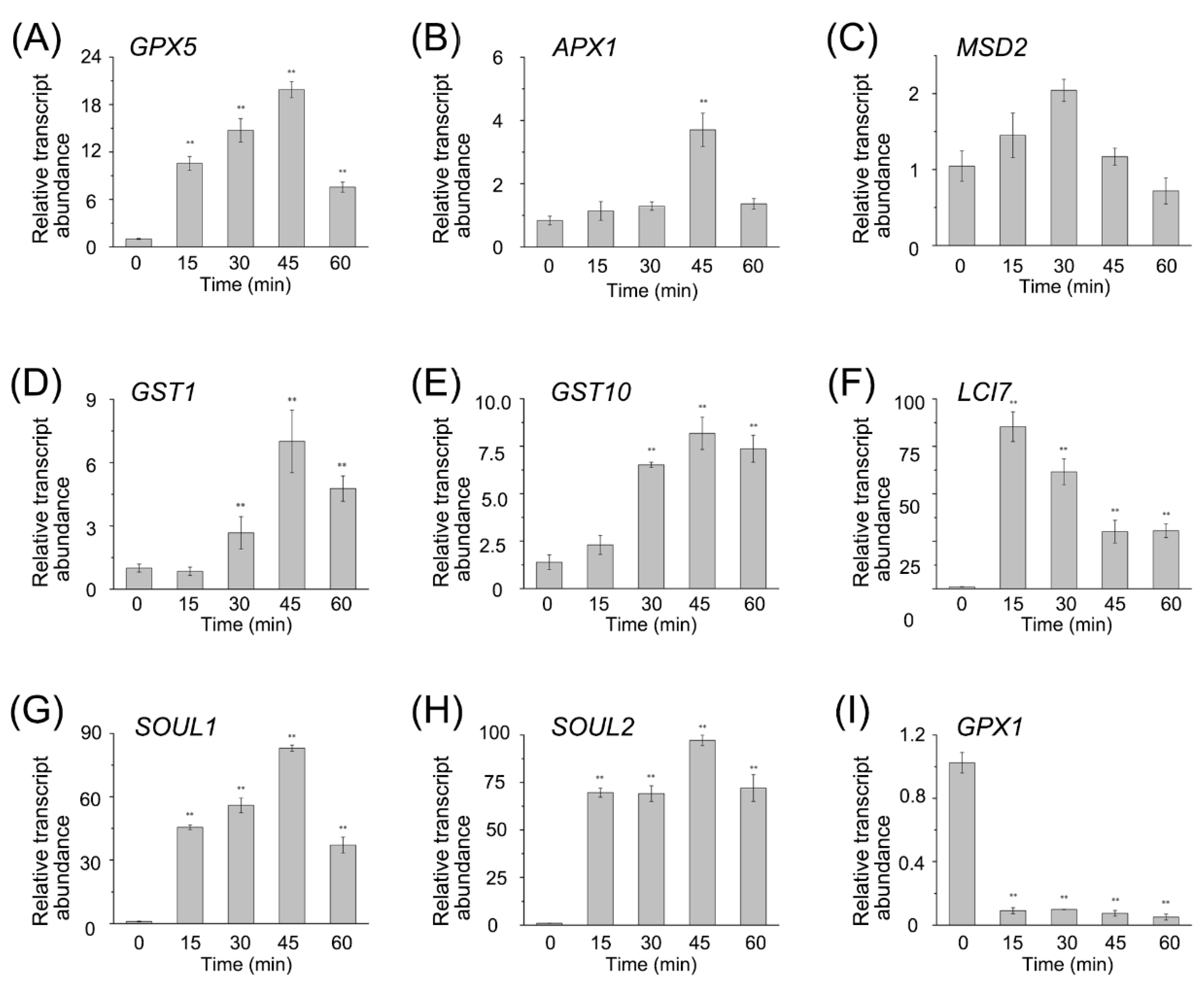
To confirm the RNA-seq data, we performed qRT-PCR on a set of DEGs, including the
GPXs,
APX1,
Mn superoxide dismutase (
MSD2),
SOUL1,
SOUL2,
LCI7, and the
GSTs (
Figure 4). The expression pattern from qRT-PCR was similar to that from RNA-seq, and the coefficient of association between them was about 0.84 (
Figure S3). Both RNA-seq data and qRT-PCR results showed that most of the transcript abundances of enzymes to detoxify ROS were upregulated. Overall, after the RB treatment, the antioxidant system was activated in CC4348 cells via the expression of related genes, providing sufficient antioxidant proteins to eliminate the
1O
2 stress generated by RB.
RNA-seq data also show that treatment with RB induced the expression of an iron-assimilating protein gene (
FEA1, Cre12.g546550) and stress-related molecular chaperones, which assist in proper protein folding. The transcript abundance of
FEA1 increased to 1703 RPKM after 30 min of stress from an initial value of 590 RPKM. Molecular chaperones also responded strongly. For example, the transcript abundance of
HSP70A increased about 3-fold, from 261 to 726 RPKM, while HSP90A increased more than 3-fold, from 275 to 901 RPKM after treatment with RB for 30 min, respectively (
Figure 3C and
Table S3). Genes encoding heat shock proteins (HSP22E, HSP22F, HSP70C, HSP70E, HSP90B, and HSP90C), CLPB3 (a ClpB chaperone belong to HSP100 family, Cre02.g090850), HOP1 (HSP70-HSP90 organizing protein), CPN60C (Chaperonin 60C), a DnaJ-like protein DNJ34, and HSP90-cochaperone (Cre07.g341550) were also all upregulated in CC4348 cells after
1O
2 stress for 30 min. This upregulation of the expression of molecular chaperones could be due to the accumulation of incorrectly folded or oxidatively damaged proteins due to oxidative stress. In support of this conclusion, we found that the transcript abundance of 17 proteasome-related genes, encode proteins involved in ubiquitination-dependent proteasome-degradation pathways, including ubiquitin-activated enzyme E1 (UBA1), ubiquitin ligase E3 (Cre12.g533750, Cre12.g500550, Cre12.g501450, and Cre11.g476250), and seven proteasome subunits, increased more than 2-fold. In addition, the transcripts of genes encoding seven proteases or metalloproteinases were upregulated.
We found 27 DEGs related to carbon metabolism, including glycolysis, the TCA cycle, and oxidative pentose phosphate pathway (OPP), and expression of 25 genes was upregulated after treatment with RB for 30 min. Four rate-limiting enzymes, a fructose-bisphosphatase (FBP1), phosphofructokinases (PFK1 and PFK2), and a pyruvate kinase (PYK4) involved in glycolysis were all induced. Notably, the PYK4 transcript increased about 5-fold to peak at 112 RPKM. Transcripts encoding another three key enzymes involved in the TCA cycle, 2-oxoglutarate dehydrogenase, citrate synthase, and isocitrate dehydrogenase, were also upregulated more than 2.5-fold. Only carbonic anhydrase (CAH8) and chitinase (Cre07.g317250) showed a different expression pattern relative to other proteins involved in carbon metabolism. On the whole, glycolysis and the TCA cycle pathway showed a rising trend, which might provide more ATP to respond to oxidative stress.
The expression of 21 transcripts related to lipid metabolism was upregulated, and only two genes were downregulated. Transcripts encoding cyclopropane fatty acid synthase (
CFA1 and
CFA2) increased in abundance by about 19-fold and 32-fold, to 217 and 70 RPKM, respectively (
Figure 3B,C,
Table S3). The transcript of a sterol-sensing 5-transmembrane protein (
SSD1) could hardly be detected under normal growth conditions, but after 30 min of RB treatment, the
SSD1 transcript reached 10 RPKM. SSD1 can function as a lipid transporter; therefore, the increase of
SSD1 transcript implied that lipid composition changed in response to oxidative stress.
Enriched classes of DEGs also included transcripts related to hormone metabolism. For example, seven enzymes involved in jasmonic acid (JA) biosynthesis were induced after RB treatment for 30 min, including Acyl-CoA oxidase (
ACO2,
ACO3, and
ACO4) and 3-hydroxyacyl-CoA dehydrogenase (
HCD1), acyl-coenzyme A thioesterase 9 (
TEH6, Cre16.g683350), lnoleate 13S-lipoxygenase (
13-LOX, Cre12.g512300). Notably,
OPR encoding 12-oxophytodienoic acid reductase (Cre03.g210513) increased in transcript abundance by about 6-fold, from 38 to 226 RPKM. The identity between 13S-lipoxygenase (13-LOX, Cre12.g512300) in
C. reinhardtii to lipoxygenase (LOX1, AT1G55020) in
Arabidopsis thaliana, 12-oxophytodienoic acid reductase (OPR, Cre03.g210513) in
C. reinhardtii to oxophytodienoate-reductase 3 (OPR3, AT2G06050) in
Arabidopsis thaliana is 36% and 46%, respectively, so we are convinced to conclude that jasmonic acid biosynthesis occurred in
C. reinhardtii. JA regulates the induced defense responses to heavy metals in the green alga
Chlorella vulgaris [
42] and cold tolerance in
Arabidopsis leaves [
43]. Therefore, we speculated that JA might also play an important role in responding to RB treatment in
C. reinhardtii.
In response to RB treatment, 94% of the genes encoding cell-structure-related proteins were downregulated. Moreover, 88% (134 DEGs) of the downregulated genes encoded flagellar associated proteins (FAPs), including Intraflagellar Transport (IFT) proteins. To further explore whether oxidative stress affected the expression of FAPs, we examined CC125 cells treated with 1 μM RB for 1 h. Because CC4348 strain is the mutant without flagella, widely used CC125 strain was used to measure the length and ratio of flagella. Up to 37% of these cells lost their flagella at 12 h. The length of flagella did not change (
Figure S4), suggesting that RB induced deflagellation, not resorption.
Overall, 1O2 activated the detoxication of ROS, stimulated increases in the abundance of HSP family proteins and ubiquitin-mediated proteolysis, upregulated carbon metabolism and lipid metabolism, and induced deflagellation.
3.4. Transcriptome Responses to 1O2 in the gpx5 Mutant
To address the function of GPX5 in response to oxidative stress, we compared the transcriptomes of the parental strain CC4348 and the
gpx5 mutant during
1O
2 stress. RNA-seq was performed on the parental strain CC4348, the
gpx5 mutant, and complemented strain L27 at 0, 30, and 60 min after the addition of 1 μM RB (
Figure 5). Using the same screening criteria, we identified 1852 DEGs in CC4348 or the
gpx5 mutant under oxidative stress. To compare the responses of CC4348 and the
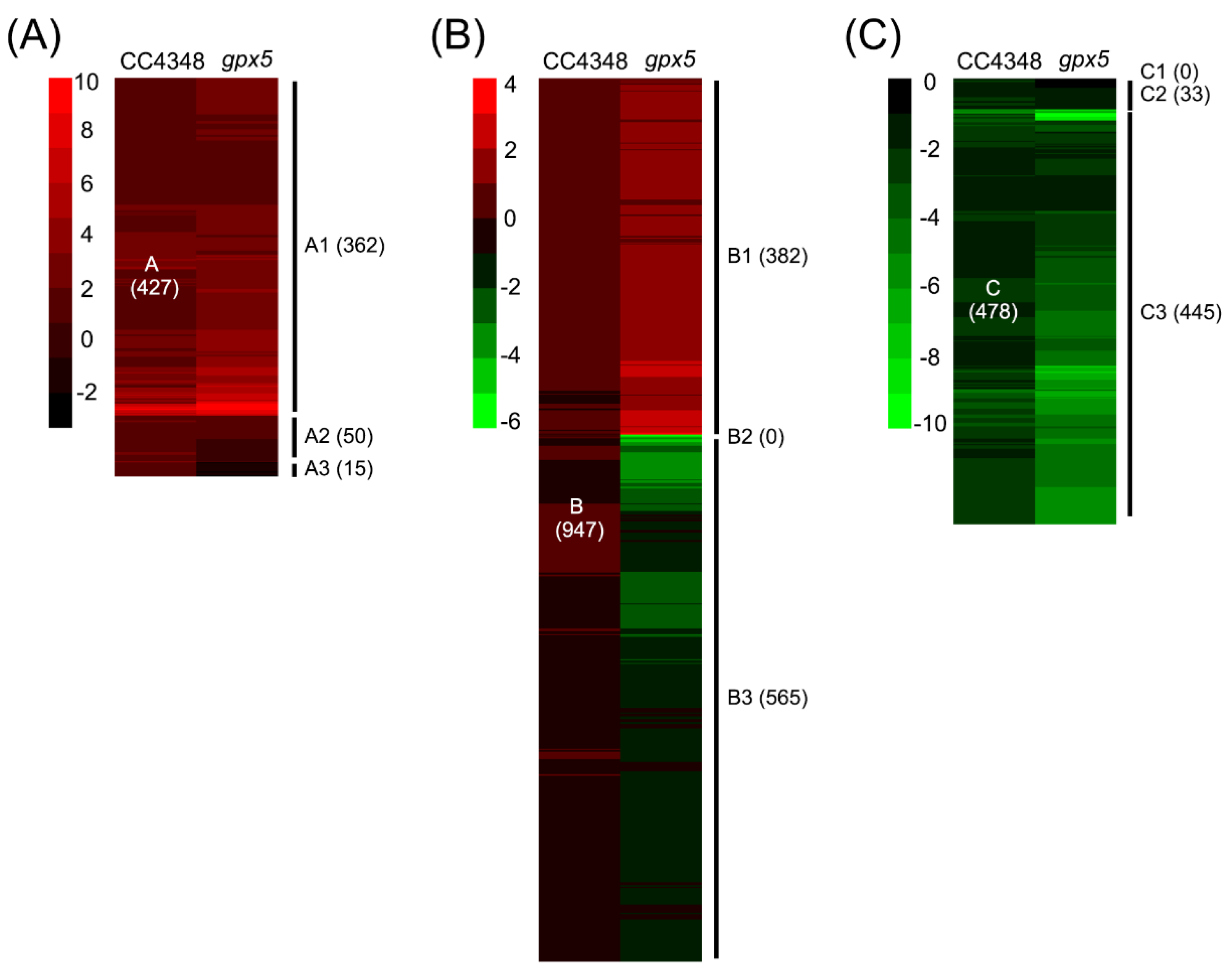
gpx5 mutant, we divided the 1852 DEGs into three categories. Group A were upregulated during the oxidative stress in CC4348 and included 947 genes. Group B did not have significantly different expression during the treatment and had 478 genes. Group C were downregulated during the response to
1O
2 in CC4348. An overview of the functions related to these transcript categories is presented in
Table 1, with an emphasis on differences between CC4348 and
gpx5 mutant cells (
Table S4).
The group A DEGs were further classified into three subgroups, A1 (362 genes, upregulated in CC4348 and
gpx5), A2 (50 genes, upregulated in CC4348, not changed in
gpx5), and A3 (15 genes, upregulated in CC4348 and downregulated in
gpx5) (
Figure 5A). The highest enriched categories of the subgroup A1 proteins include ROS detoxification, carbon metabolism, and ubiquitin-dependent proteolysis. In subgroup A1, 31 transcripts encoded proteins regulating redox homeostasis, including APX1, GSR1, GST6, GST10, VTC2, TRXs, and SRX1. These antioxidases or antioxidant proteins responded to oxidative stress independent of GPX5.
Although the transcript abundance of most genes related to carbon metabolism was lower in the gpx5 mutant than that in CC4348 under normal growth conditions, transcripts encoding proteins related to glycolysis and the oxidative pentose phosphate pathway (OPP), such as FBP1, PFK1, PFK2, and transaldolases (TAL1, TAL2) from subgroup A1, were upregulated in gpx5 in the same pattern as in CC4348 after RB treatment. In particular, the TAL1 transcript increased from 26 to 194 RPKM in gpx5 and from 89 to 250 RPKM in CC4348. Increasing glycolysis metabolism and OPP would likely produce more NADPH to counteract the oxidative environment in cells.
Genes from subgroups A2 and A3 (upregulated in CC4348 and unchanged or downregulated in gpx5) are mainly involved in chlorophyll metabolism or related to redox or transport. Eight transcripts encoding proteins involved in chlorophyll synthesis from subgroup A2 were significantly increased in abundance in CC4348, whereas those mostly remained constant in the gpx5 mutant after RB treatment. Magnesium chelatase subunit transcripts (CHLD, CHLI1, and CHLI2) were upregulated by more than 2-fold (peak more than 50 RPKM) in CC4348 after treatment, but did not significantly change in the gpx5 mutant. Subgroup A3 had only one transcript related to chlorophyll synthesis, CPX1 (coproporphyrinogen III oxidase), which was upregulated in CC4348 but downregulated in the gpx5 mutant. These observations suggested that the chlorophyll content would increase in CC4348 but not in gpx5 after RB treatment.
Five transcripts from subgroup A2 and 3 transcripts from subgroup A3 encoded putative transporters, including transporters in vesicular trafficking (Cre04.g224800 and Cre09.g394954), mitochondrial oxoglutarate/malate carrier protein (Cre11.g467535), sodium-exporting ATPase (Cre17.g744447), and major facilitator superfamily transporter (MFT10, Cre02.g095076) from subgroup A2, and sodium/bile acid cotransporter 7 (Cre02.g095086), MFT26 (Cre12.g512200), and a solute carrier family 35 member (Cre06.g286200) from A3.
We also observed that the abundance of transcripts encoding thioredoxin peroxidase (Cre16.g654250) from subgroup A2 and NRX2 from subgroup A3 increased in CC4348 under oxidative stress, but not in the gpx5 mutant. The transcript for a 60-kDa SS-A/Ro ribonucleoprotein (Cre17.g725750) from subgroup A3 increased in abundance, from 35 to 93 RPKM in CC4348 after RB treatment for 30 min, but decreased from 136 to 37 RPKM in the gpx5 under the same conditions. This RNA-binding protein is differentially expressed depending on GPX5 under oxidative stress, suggesting that it regulates the downstream response.
Genes in group B were similarly classified into subgroups, according to their response in the
gpx5 mutant under oxidative stress. Among them, 382 genes, which were designated as B1, were upregulated in the
gpx5 mutant, and 565 genes, designated B3, were downregulated in the
gpx5 mutant (
Figure 5B). Therefore, genes in subgroup B1 were specifically upregulated in the
gpx5 mutant, and genes in subgroup B3 were specifically downregulated in the
gpx5 mutant.
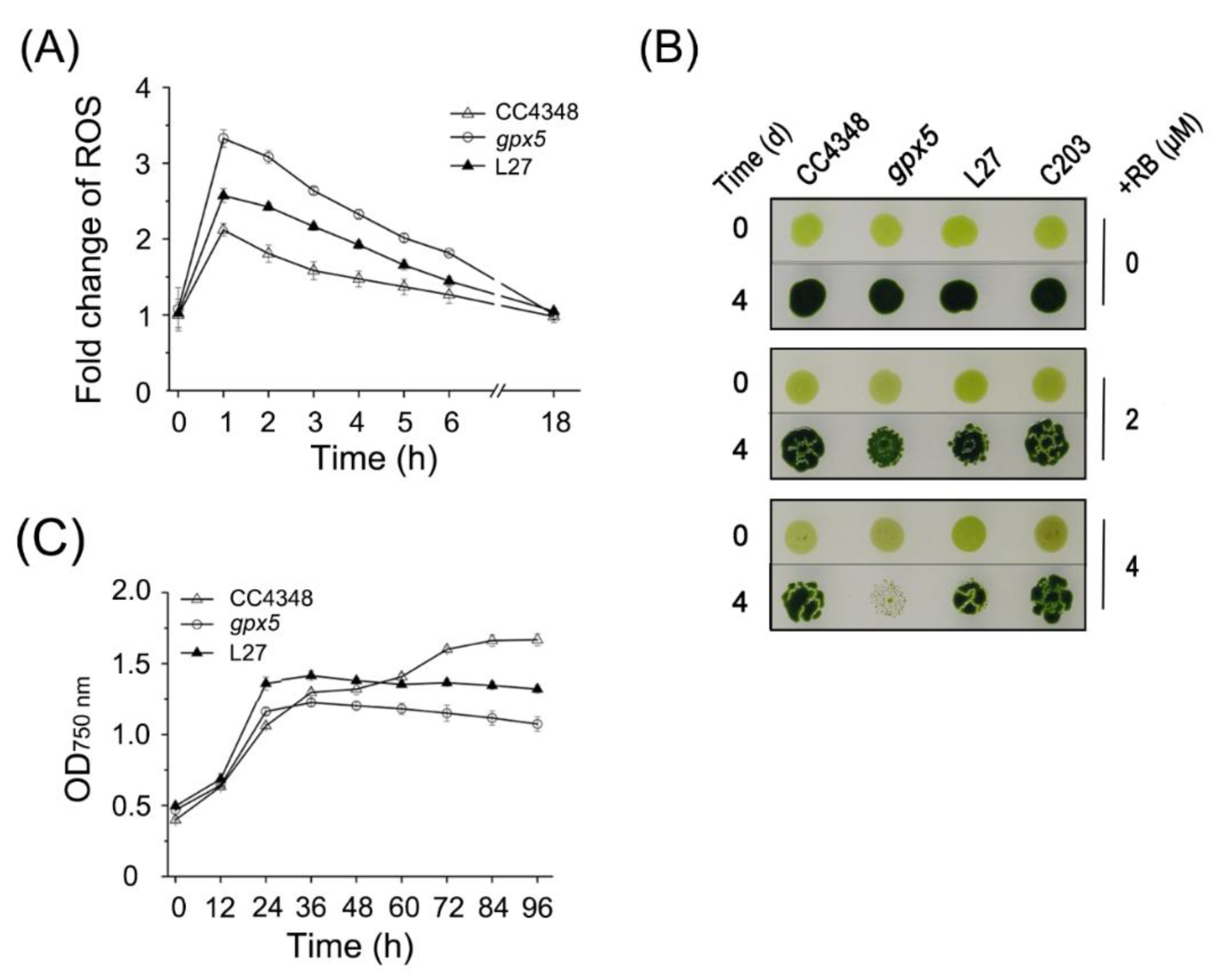
Unlike the parental strain CC4348, the
gpx5 cells had high concentrations of ROS after RB treatment (
Figure 1A). The high concentration of ROS induced transcripts from subgroup B1, which are mainly associated with the mETC, carbon metabolism, protein metabolism, and endosomal sorting complex required for transport (ESCRT). The transcript abundance of genes encoding cytochrome c oxidase subunits (
COX2A and
COX3), cytochrome c oxidase assembly factor (
SCO1), ubiquinone cytochrome c oxidoreductase subunit (
QCR1), and eight mitochondrial ATP synthase subunits in the mETC remained stable in CC4348, but increased by 2- to 5-fold in the
gpx5 mutant after oxidative stress treatment. Besides, we found a type II NAD(P)H dehydrogenase (NDA1), an alternative oxidase (AOX1), complex IV (SCO1 and PET191) upregulated, and this fits well the data published by Sabeeha S. Merchant [
44]. They showed transcripts encoding proteins of the most mETC genes not changed and the O
2 consumption decreased. These results suggested that ATP produced by respiration is in smaller amounts in stress conditions.
Transcripts encoding proteins involved in carbon metabolism, especially the TCA cycle and starch metabolism, were also enriched in subgroup B1. Indeed, 12 transcripts encoding proteins involved in the TCA, phosphoenolpyruvate carboxykinase (PCK1), mitochondrial pyruvate dehydrogenase complex alpha subunit (PDC1), isocitrate dehydrogenase (IDH1 and IDH2), Dihydrolipoamide succinyltransferase (OGD2), succinyl-CoA ligase (SCL1 and SCL2), succinate dehydrogenase (SDH1 and SDH3), fumarase (FUM1), and NAD-dependent malate dehydrogenase (MDH3 and MDH4) were significantly increased only in the gpx5 mutant after RB treatment for 30 min. In addition, OGD1, CIS1, and IDH3 increased in the gpx5 mutant and CC4348. The upregulation of the TCA cycle, along with increased mETC activity, likely supply more energy to respond to oxidative stress in the gpx5 mutant.
A comparison of
gpx5 and CC4348 also showed that transcripts related to ubiquitin-mediated proteolysis were induced after oxidative stress both in CC4348 and
gpx5 cells, including 30 transcripts from subgroup A1 and 33 transcripts from subgroup B1. Almost all transcripts related to the ubiquitin–proteasome system increased in abundance during the 60-min oxidative stress treatment in CC4348 (
Figure S5 and Table S5). Under the control conditions (0 min), the abundance of these transcripts was lower in the
gpx5 mutant than that in CC4348. After 60 min of RB treatment, these transcripts showed a dramatic upregulation in the
gpx5 mutant, significantly larger than that of CC4348. Before the treatment (0 min), the abundances of these transcripts were similar in CC4348 and
gpx5, but they increased significantly after addition of RB in L27, and the increase was between that of CC4348 and
gpx5 cells. This increase suggested that ROS stimulated the upregulation of transcripts encoding proteasome-related subunits. As ROS concentration increased, protein stress increased, resulting in upregulation of the ubiquitin–proteasome system.
Oxidative stress also induced 11 transcripts encoding proteins in the secretory pathway or vacuolar assembly/sorting proteins (VPS2A, VPS26, VPS46, and VPS60) from subgroup B1. These transcripts increased in abundance specially in the gpx5 mutant after RB treatment. These data suggest that high ROS levels accelerated the transport and exchange of metabolites between cells.
Transcripts from subgroup B3 genes were specifically downregulated in the gpx5 mutant, including ten adenylate cyclase or guanylate cyclase subunit transcripts and five 3’,5’-cyclic nucleotide phosphodiesterase transcripts, whereas there was no significant difference in CC4348. Adenylate cyclase and guanylate cyclase catalyze the formation of cAMP and cGMP, respectively. As secondary messengers, cAMP or cGMP activate kinases. The high concentration of ROS could inhibit some cAMP or cGMP signaling pathways. In addition, subgroup B3 included transcripts encoding Mitogen-Activated Protein Kinase (MAPK) signaling pathway factors. For example, MAPKKK7 (Mitogen-Activated Protein Kinase Kinase Kinase) was specifically downregulated, from 19 to 9 RPKM in the gpx5 mutant, but its expression remained stable at about 16 RPKM in CC4348. ROS also affected the calcium signal as mediated by calmodulin. Transcripts encoding calmodulins (Cre03.g210177 and Cre03.g170200), a Ca2+-transporting ATPase (Cre12.g505350), and a calcium-binding protein (Cre16.g680500) from subgroup B3 were specially downregulated in the gpx5 mutant but showed no significant difference in CC4348. This suggests that signal transduction including cAMP or cGMP, MAPK pathways, and Ca2+ signaling were inhibited in response to high concentrations of ROS.
Two iron transporter genes, IRT2, encoding an iron-responsive ZIP family transporter (Cre12.g530350) and FTR1, encoding an iron transporter (Cre03.g192050), responded to 1O2 specially in the gpx5 mutant. We speculated that iron might respond to the cellular redox state or deliver a redox signal to other ROS sensors.
Similarly, group C genes, which were downregulated in CC4348, were classified into subgroups. Interestingly, none of the genes were upregulated in the
gpx5 mutant (group C1); 33 genes, designated C2, were not significantly changed in the
gpx5 mutant; and 445 genes, designated C3, were downregulated in both
gpx5 and CC4348 (
Figure 5C).
The C2 genes encoded proteins involved in nucleotide metabolism and regulation of transcription. Transcripts of a histone H2B variant (HBV1), ATP-dependent RNA helicases (HEL12, HEL34, and HEL56), DNA-directed RNA polymerase I subunit (RPA12), and transcription initiation factor TFIID subunit 10 (Cre16.g657000) were downregulated more than 2-fold in CC4348, but remained stable in the gpx5 mutant under oxidative stress. These downregulated transcripts included some transcription factors or transcription regulators, including protein with chromosome condensation (RCC1) repeat domain (Cre12.g528350), ribosomal N-lysine methyltransferase 3 (Cre12.g541777), and leucine zipper transcription factors (Cre06.g252000 and Cre12.g490950). These transcription factors were specifically inhibited in CC4348, which might be related to GPX5 protein or low ROS concentrations.
The expression of subgroup C3 genes was downregulated both in the
gpx5 mutant and CC4348 under oxidative stress. In subgroup C3, 178 FAP transcripts were rapidly downregulated under oxidative stress in the
gpx5 mutant and CC348. These results further indicated that oxidative stress inhibits the expression of flagella-related genes, and more ROS induced deflagellation in
C. reinhardtii (
Figure 3D). In addition, the transcripts of four genes encoding selenoproteins,
GPX1,
NTR1,
SELW1, and
SELU1, from subgroup C3 were significantly downregulated in CC4348 and
gpx5 cells under oxidative stress. The relationship between selenoproteins and oxidative stress remains to be explored.
By comparing the expression of these 1852 DEGs in CC4348 and L27, we found that there were high similar responses between CC4348 and the complementary strain L27. Among 322 upregulated DEGs in CC4348 after treatment for 30 min, roughly 79% (253) overlapped with induced DEGs in L27. Among 386 downregulated DEGs in CC4348 after treatment for 30 min, roughly 88% (341) overlapped with depressed DEGs in L27. Overall, about 84% (594 out of 708) of DEGs in CC4348 were overlapped with that in L27 after treatment for 30 min (
Figure S6 and Table S4).
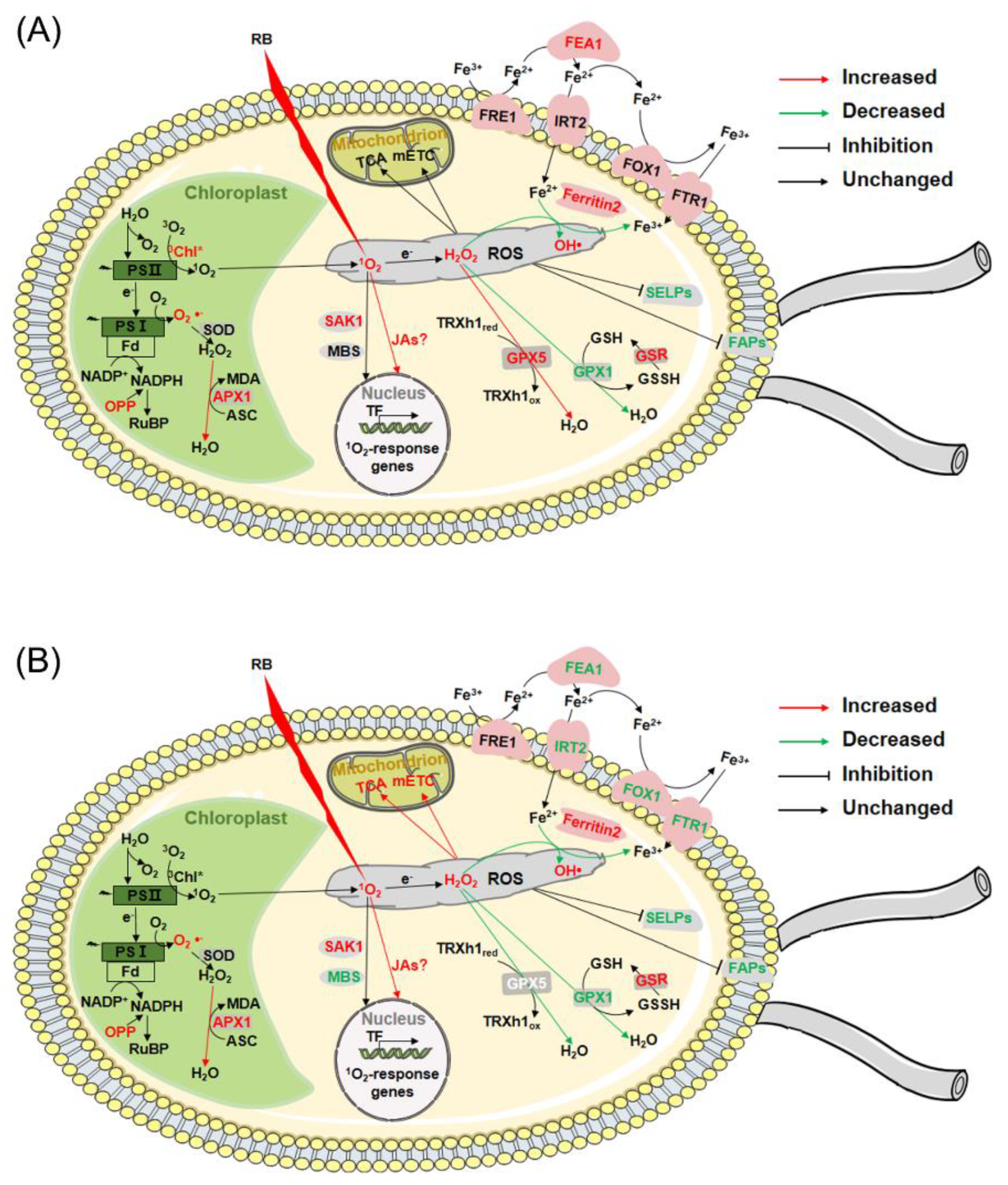
In brief, after RB treatment, CC4348 and the gpx5 mutant shared some common pathways to respond to oxidative stress showed, including upregulated ROS detoxification and ubiquitin-mediated proteolysis (subgroup A1), and inhibited the expression of cell structure related proteins and selenoproteins (subgroup C3). When GPX5 protein was deficient, some specific responses occurred, such as accelerated the TCA cycle and mETC in mitochondria to supply more ATP (subgroup B1), repressed genes related to chlorophyll metabolism and photosynthesis (subgroup A2 and A3), and downregulated iron transporters (subgroup B3).
3.5. Effect of 1O2 on Photosynthesis
Previous studies on terrestrial plants and photosynthetic microorganisms have shown that hydrogen peroxide downregulates the expression of genes encoding photosynthesis-related proteins [
45,
46,
47]. To further understand the effects of
1O
2 on the expression of photosynthesis-related genes, we analyzed the transcript changes of LHC genes. The transcript level of
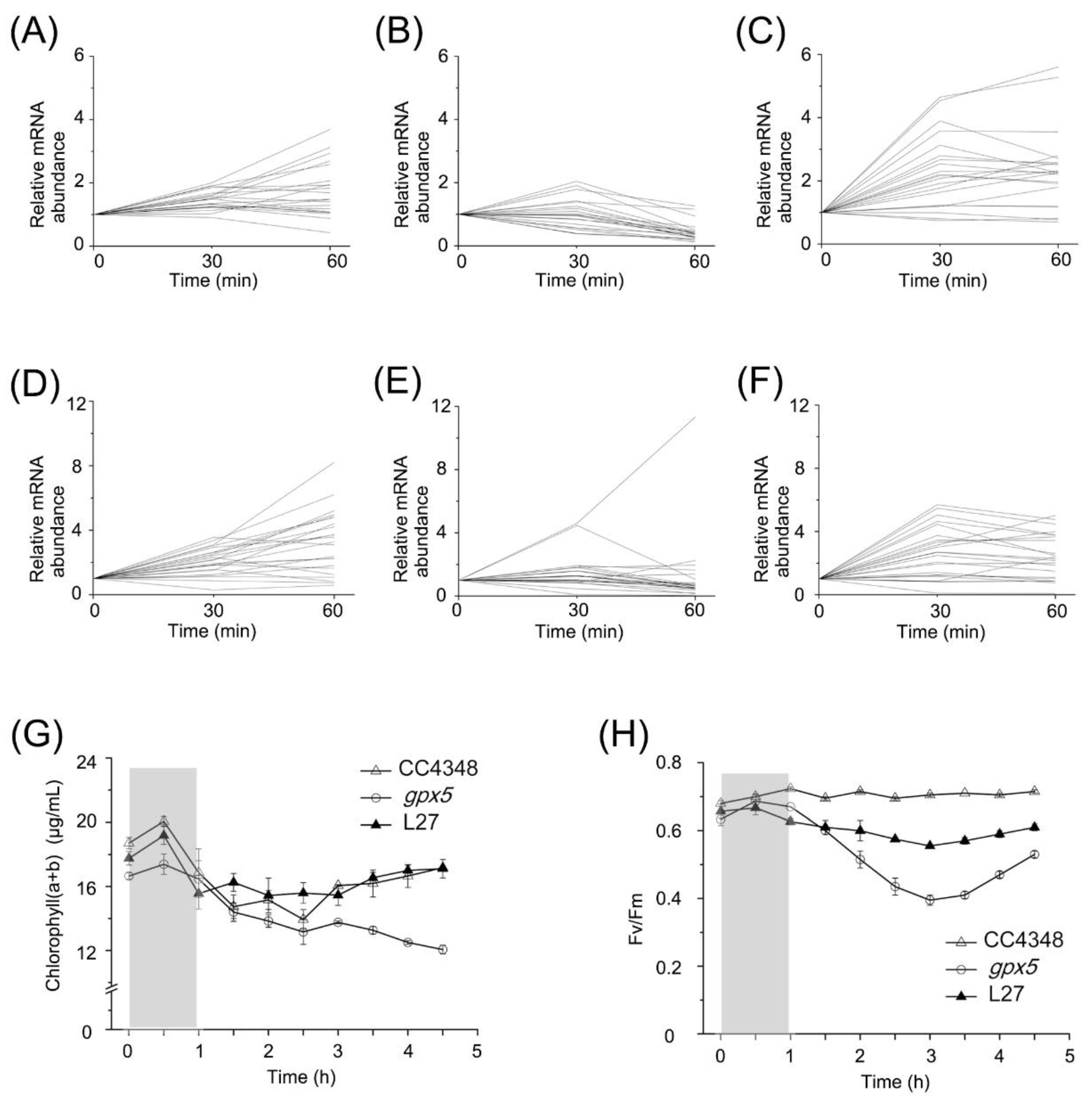
LHCs in CC4348 increased by two- to four-fold within 1 h after RB treatment. However, they fluctuated over a two-fold range during oxidative stress in the
gpx5 mutant, but increased significantly in L27, even more than in CC4348 after RB treatment for 30 min (
Figure 6A–C and
Table S6).
RNA-seq data of transcripts encoding proteins involved in chlorophyll metabolism in CC4348,
gpx5, and L27 strains were also analyzed at 0, 30, and 60 min after RB treatment (
Figure 6D–F). The expression of chlorophyll-metabolism-related genes in CC4348 showed a continuous trend of upregulation within 60 min of oxidative stress, whereas those in the
gpx5 mutant showed an upward trend in expression within the first 30 min and remained stable in the following 30 min, except a chlorophyll synthase gene (
CHLG, Cre06.g294750) and a delta-aminolevulinic acid dehydratase gene (
ALAD1, Cre02.g091050) (
Figure 6E). Most of these transcripts were upregulated by less than 6-fold after 30 min and then remained stable abundance in L27. In CC4348, the transcript abundance of
CHLG was continuously increased from 12 RPKM after RB treatment and reached a peak of 45 RPKM after 60 min; in the
gpx5 mutant,
CHLG reached a peak of 11 RPKM after 30 min and then decreased to 3 RPKM. Although the expression of
CHLG in the
gpx5 mutant increased by 4.5-fold, the overall transcript abundance was low. CHLG catalyzes the production of chlorophyll from chlorophyllide. The relative low abundance of
CHLG transcript affect the final synthesis of pigments. In addition, the
ALAD1 transcript increased more than 6-fold (peak at 45 RPKM) after RB treatment for 60 min in CC4348, and increased about 11-fold (peak at 55 RPKM) in
gpx5. The expression pattern of
ALAD1 in L27 was similar with that in CC4348.
ALAD1 was more strongly induced by
1O
2 than other genes in the chlorophyll synthesis pathway, suggesting that it is important for this response.
To determine the effect of oxidative stress on the photosynthetic efficiency of CC4348 and the
gpx5 mutant, we measured chlorophyll pigment contents and photosynthetic efficiency every 30 min, during 1 h of treatment with 1 µM RB and another 3.5 h after the treatment was terminated.
Figure 6G shows the total chlorophyll
a and
b contents. Consistent with the mRNA abundance of genes related to chlorophyll metabolism, the chlorophyll content of CC4348 increased by about 7% after a 30-min treatment and then returned to 91% of the initial level after the 3.5 h recovery. The chlorophyll in the
gpx5 mutant increased by only 4% in the first 30 min of treatment but then decreased continuously after removal of RB. The pigment content decreased to 72% of the initial state in the
gpx5 mutant at 3.5 h after termination of the RB treatment. The pattern of change of pigment content in L27 was similar to that of CC348, in which chlorophyll increased by 8% after 30 min of treatment and then decreased to 96% of the initial level. Therefore, we speculated that a low ROS concentration may stimulate the transient synthesis of chlorophyll, whereas a high ROS concentration may inhibit chlorophyll metabolism.
The maximum efficiency of PSII (
Figure 6H) was also measured. The photosynthetic efficiency of PSII averaged around 0.7 in CC4348 throughout the experiment. However, at the initial time point, the maximum efficiency was only 0.63 in the
gpx5 mutant, but this increased to 0.68 after 30 min of RB treatment. At 3 h after the RB was removed, the maximum efficiency of PSII had declined to its lowest value, 0.4, in the
gpx5 mutant. At 3.5 h after the RB was removed, the maximum efficiency slowly recovered to 0.53 in the mutant, which was still lower than its initial level. The maximum efficiency of PSII of L27 was similar to that of CC348, but its value was slightly lower (about 0.6). Oxidative stress had little effect on the photosynthetic efficiency of CC348, but it had a negative effect on the
gpx5 mutant. After treatment with RB, ROS accumulated in
gpx5 cells. Therefore, a high concentration of ROS might inhibit photosynthetic efficiency. After the removal of oxidative stress, the ROS level in cells gradually decreased and the photosynthetic efficiency recovered accordingly. These results indicated that a small amount of ROS had little effect on photosynthetic efficiency, but that a high concentration of ROS inhibited photosynthetic efficiency.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}