Satellitome Analysis in the Ladybird Beetle Hippodamia variegata (Coleoptera, Coccinellidae)

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
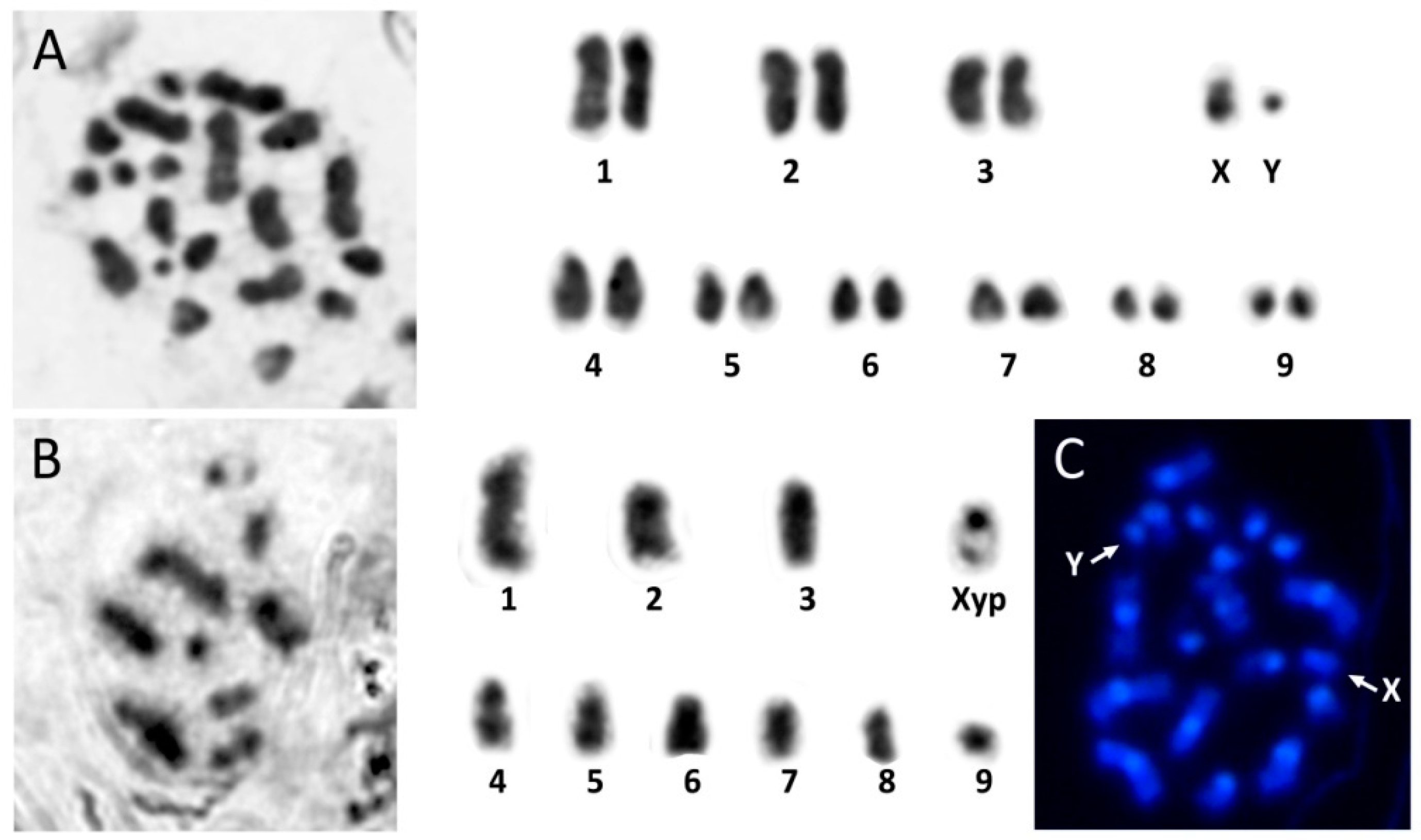
2.1. Sampling, Chromosome Preparation and DNA Extraction
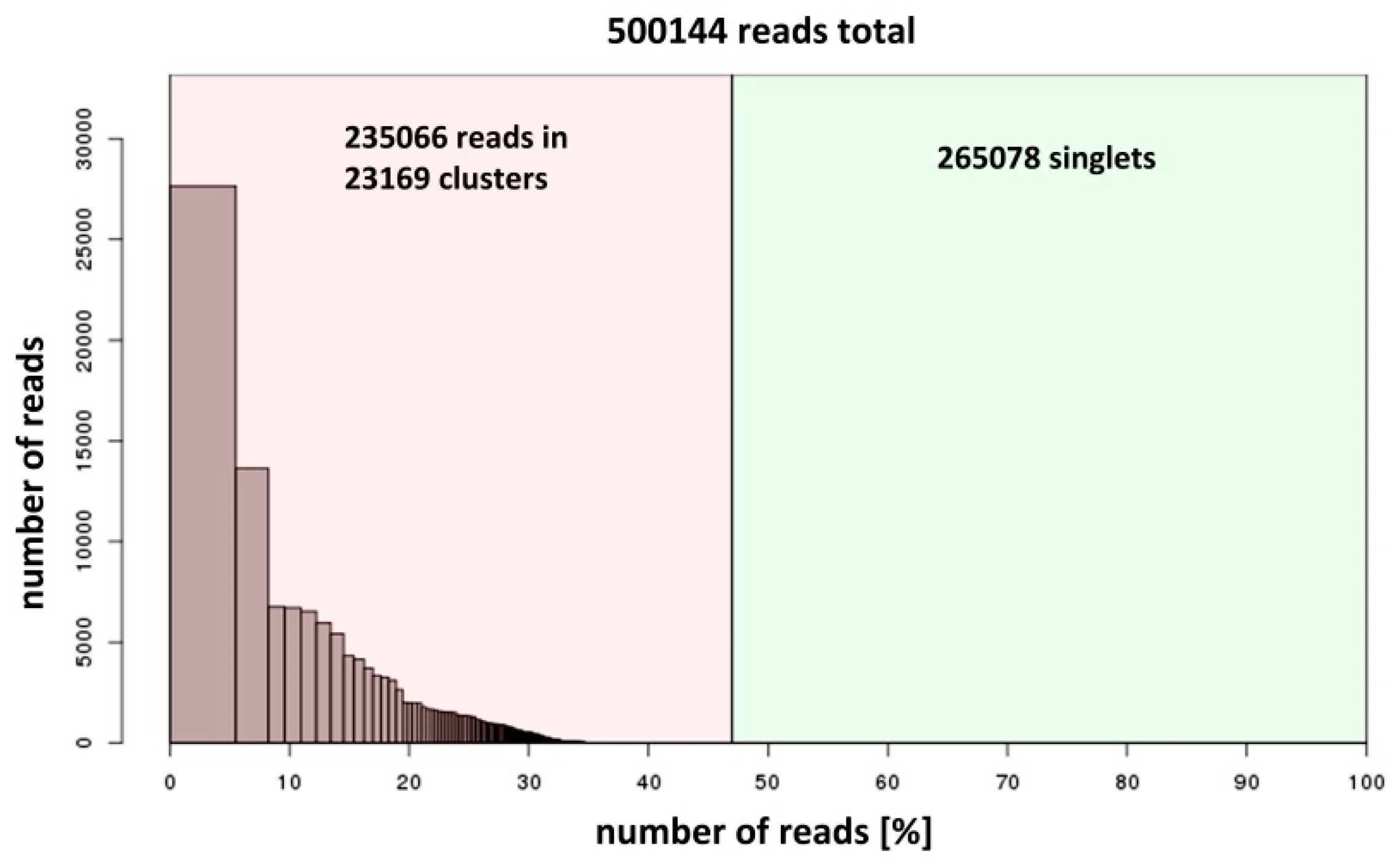
2.2. DNA Sequencing and Data Analysis
2.3. PCR Amplification, Sequences Cloning and Cytogenetic Mapping
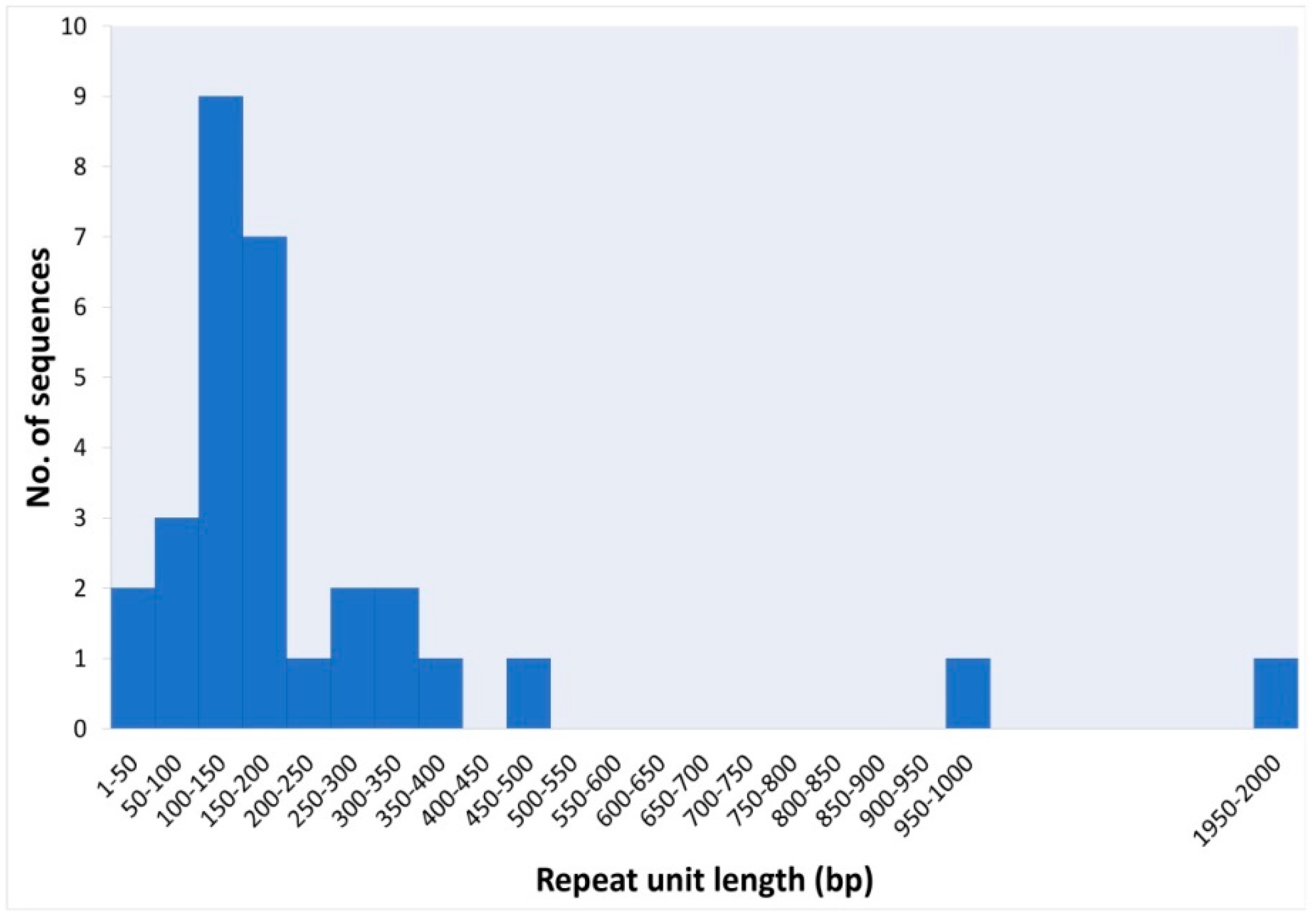
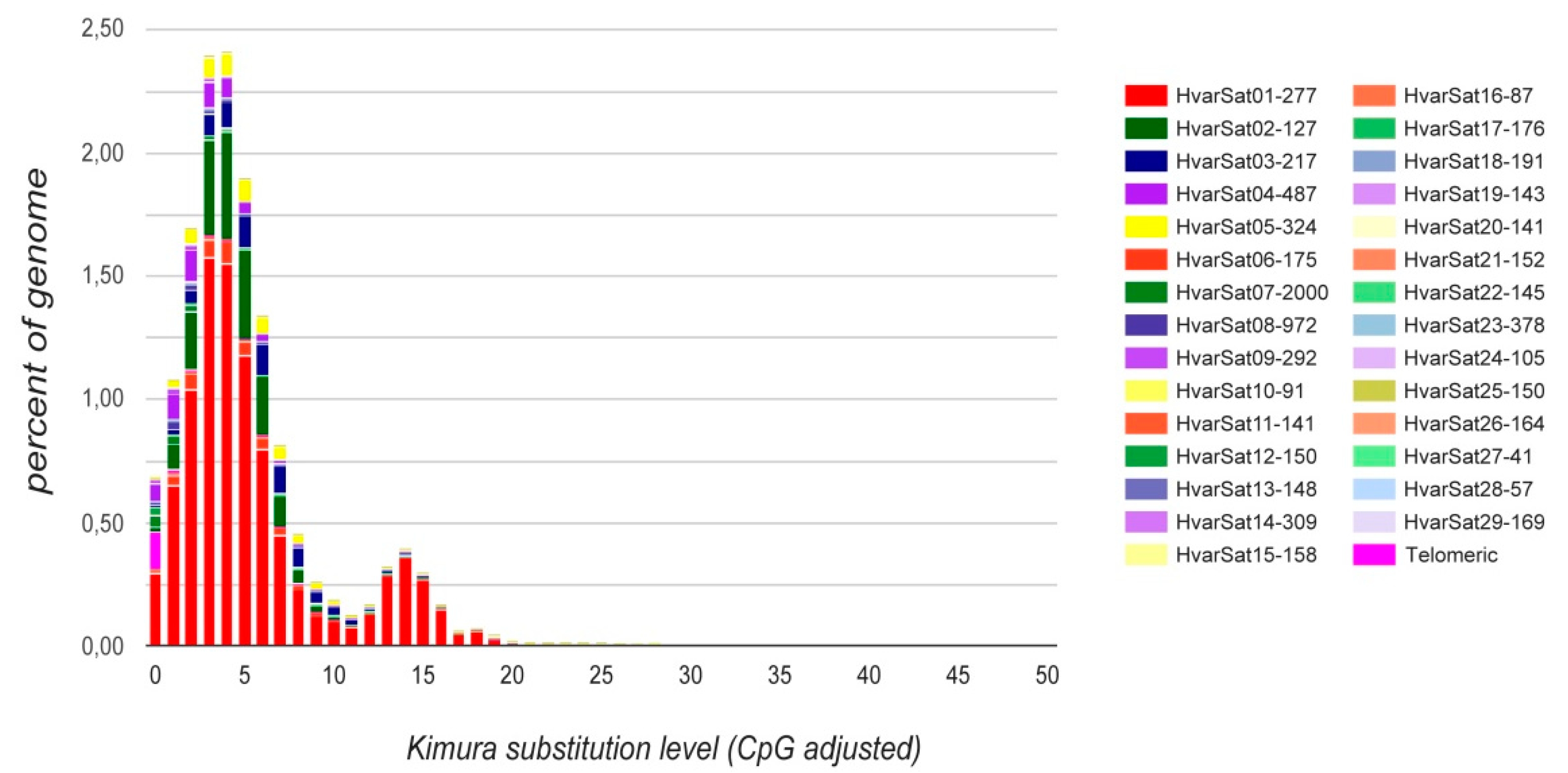
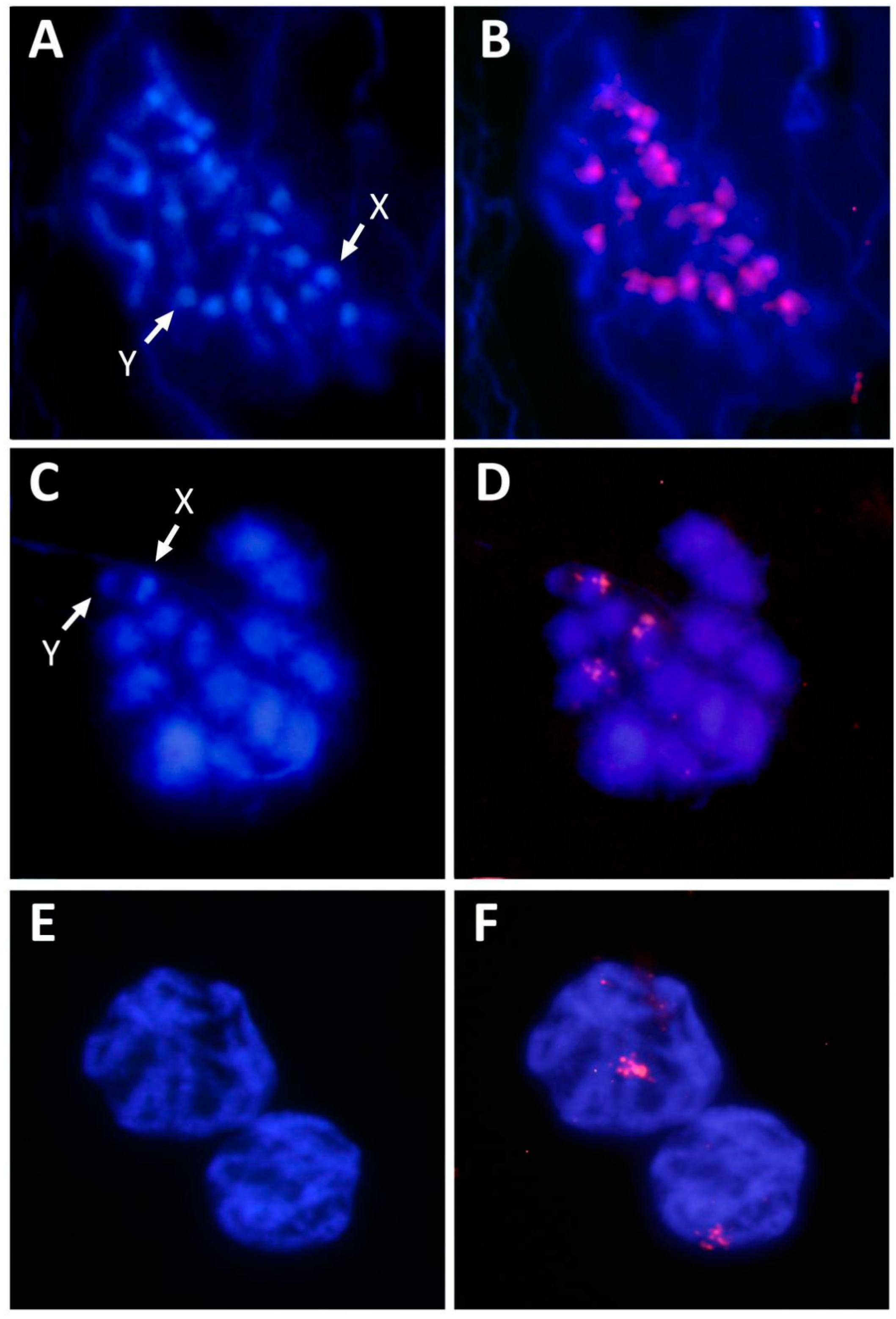
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Garrido-Ramos, M.A. Satellite DNA: An evolving topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef]
- Kit, S. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J. Mol. Biol. 1961, 3, 711–716. [Google Scholar] [CrossRef]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Ugarković, D.; Plohl, M. Variation in satellite DNA profiles-causes and effects. EMBO J. 2002, 21, 5955–5959. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.; Petitpierre, E.; Juan, C. Characterization of the heterochromatin of the darkling beetle Misolampus goudoti: Cloning of two satellite DNA families and digestion of chromosomes with restriction enzymes. Hereditas 1993, 119, 179–185. [Google Scholar] [CrossRef]
- Palomeque, T.; Lorite, P. Satellite DNA in insects: A review. Heredity 2008, 100, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Neumann, P.; Novák, P.; Jiang, J. Global sequence characterization of rice centromeric satellite based on oligomer frequency analysis in large-scale sequencing data. Bioinformatics 2010, 26, 2101–2108. [Google Scholar] [CrossRef]
- Shapiro, J.A.; von Sternberg, R. Why repetitive DNA is essential to genome function. Biol. Rev. 2005, 80, 227–250. [Google Scholar] [CrossRef]
- Hughes, S.E.; Hawley, R.S. Heterochromatin: A rapidly evolving species barrier. PLoS Biol. 2009, 7, e1000233. [Google Scholar] [CrossRef]
- Ferree, P.M.; Prasad, S. How can satellite DNA divergence cause reproductive isolation? Let us count the chromosomal ways. Genet. Res. Int. 2012, 430136. [Google Scholar] [CrossRef]
- Plohl, M.; Luchetti, A.; Meštrović, N.; Mantovani, B. Satellite DNAs between selfishness and functionality: Structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 2008, 409, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Goyal, V. Repetitive sequences in plant nuclear DNA: Types, distribution, evolution and function. Genom. Proteom. Bioinf. 2014, 12, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. Satellite DNA in plants: More than just rubbish. Cytogenet. Genome Res. 2015, 146, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Lorite, P.; Torres, M.I.; Palomeque, T. Characterization of two unrelated satellite DNA families in the Colorado potato beetle Leptinotarsa decemlineata (Coleoptera, Chrysomelidae). Bull. Entomol. Res. 2013, 103, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Mora, P.; Vela, J.; Palomeque, T.; Sánchez, A.; Panzera, F.; Lorite, P. Comparative analysis of repetitive DNA between the main vectors of Chagas disease: Triatoma infestans and Rhodnius prolixus. Int. J. Mol. Sci. 2018, 19, 1277. [Google Scholar] [CrossRef]
- Pita, S.; Panzera, F.; Mora, P.; Vela, J.; Cuadrado, A.; Sánchez, A.; Palomeque, T.; Lorite, P. Comparative repeatome analysis on Triatoma infestans Andean and non-Andean lineages, main vector of Chagas disease. PLoS ONE 2017, 12, e0181635. [Google Scholar] [CrossRef]
- Camacho, J.P.; Ruiz-Ruano, F.J.; Martín-Blázquez, R.; López-León, M.D.; Cabrero, J.; Lorite, P.; Cabral-de-Mello, D.C.; Bakkali, M. A step to the gigantic genome of the desert locust: Chromosome sizes and repeated DNAs. Chromosoma 2015, 124, 263–275. [Google Scholar] [CrossRef]
- Maumus, F.; Quesneville, H. Deep investigation of Arabidopsis thaliana junk DNA reveals a continuum between repetitive elements and genomic dark matter. PLoS ONE 2014, 9, e94101. [Google Scholar] [CrossRef]
- Ruiz-Ruano, F.J.; López-León, M.D.; Cabrero, J.; Camacho, J.P.M. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci. Rep. 2016, 6, 28333. [Google Scholar] [CrossRef]
- Altemose, N.; Miga, K.H.; Maggioni, M.; Willard, H.F. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput. Biol. 2014, 10, e1003628. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExporer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef]
- Novák, P.; Ávila-Robledillo, L.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef] [PubMed]
- Feliciello, I.; Chinali, G.; Ugarković, D. Structure and population dynamics of the major satellite DNA in the red flour beetle Tribolium castaneum. Genetica 2011, 139, 999–1008. [Google Scholar] [CrossRef]
- Feliciello, I.; Akrap, I.; Brajković, J.; Zlatar, I.; Ugarković, D. Satellite DNA as a driver of population divergence in the red flour beetle Tribolium castaneum. Genome Biol. Evol. 2015, 7, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruano, F.J.; Castillo-Martínez, J.; Cabrero, R.; Gómez, J.; Camacho, J.P.M.; López-León, M.D. High-throughput analysis of satellite DNA in the grasshopper Pyrgomorpha conica reveals abundance of homologous and heterologous higher-order repeats. Chromosoma 2018, 127, 323–340. [Google Scholar] [CrossRef]
- Palacios-Gimenez, O.M.; Dias, G.B.; de Lima, L.G.; Kuhn, G.; Ramos, É.; Martins, C.; Cabral-de-Mello, D.C. High-throughput analysis of the satellitome revealed enormous diversity of satellite DNAs in the neo-Y chromosome of the cricket Eneoptera surinamensis. Sci. Rep. 2017, 7, 6422. [Google Scholar] [CrossRef]
- Palacios-Gimenez, O.M.; Bardella, V.B.; Lemos, B.; Cabral-de-Mello, D.C. Satellite DNAs are conserved and differentially transcribed among Gryllus cricket species. DNA Res. 2018, 25, 137–147. [Google Scholar] [CrossRef]
- Bouchard, P.; Grebennikov, V.V.; Smith, A.B.T.; Douglas, H. Biodiversity of Coleoptera. In Insect Biodiversity: Science and Society; Foottit, R.G., Adler, P.H., Eds.; Blackwell Publishing: Oxford, UK, 2009; pp. 265–301. [Google Scholar] [CrossRef]
- Nedvěd, O.; Kovář, I. Phylogeny and classification. In Ecology and Behaviour of the Ladybird Beetles (Coccinellidae); Hodek, I., van Emden, H.F., Honěk, A., Eds.; Blackwell Publishing Ltd.: Chichester, UK, 2012; pp. 1–12. [Google Scholar] [CrossRef]
- Dixon, A.F.G. Insect Predator-Prey Dynamics; Ladybird Beetles and Biological Control; Cambridge University Press: Cambridge, UK, 2005; p. 257. [Google Scholar]
- Khan, A.A.; Zaki, F.A.; Khan, Z.H.; Mir, R.A. Biodiversity of predacious ladybird beetles (Coleoptera: Coccinellidae) in Kashmir. J. Biol. Control 2009, 23, 43–47. [Google Scholar] [CrossRef]
- Hodek, I.; Evans, E.W. Food relationships. In Ecology and Behaviour of the Ladybird Beetles (Coccinellidae); Hodek, I., van Emden, H.F., Honěk, A., Eds.; Wiley-Blackwell: Chichester, UK, 2012; pp. 141–274. [Google Scholar] [CrossRef]
- Eliopoulos, P.A.; Kontodimas, D.C.; Stathas, G.J. Temperature-dependent development of Chilocorus bipustulatus (Coleoptera: Coccinellidae). Environ. Entomol. 2010, 39, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Hao, Y.N.; Riddick, E.W.; Liu, T.X. Factitious prey and artificial diets for predatory lady beetles: Current situation, obstacles, and approaches for improvement: A review. Biocontrol Sci. Tech. 2017, 27, 601–619. [Google Scholar] [CrossRef]
- Obrycki, J.J.; Kring, T.J. Predaceous Coccinellidae in biological control. Annu. Rev. Entomol. 1998, 43, 295–321. [Google Scholar] [CrossRef] [PubMed]
- Cotes, B.; Campos, M.; Pascual, F.; Ruano, F. The ladybeetle community (Coleoptera: Coccinellidae) in Southern olive agroecosystems of Spain. Environ. Entomol. 2010, 39, 79–87. [Google Scholar] [CrossRef]
- Iperti, G. Biodiversity of predaceous coccinellidae in relation to bioindication and economic importance. Agric. Ecosyst. Environ. 1999, 74, 323–342. [Google Scholar] [CrossRef]
- Rondoni, G.; Ielo, F.; Ricci, C.; Conti, E. Intraguild predation responses in two aphidophagous Coccinellids identify differences among juvenile stages and aphid densities. Insects 2014, 5, 974–983. [Google Scholar] [CrossRef]
- Skouras, P.J.; Stathas, G.J. Development, growth and body weight of Hippodamia variegata fed Aphis fabae in the laboratory. Bull. Insect. 2015, 68, 193–198. [Google Scholar]
- Franzmann, B.A. Hippodamia variegata (Goeze) (Coleoptera: Coccinellidae), a predacious ladybird new in Australia. Aust. J. Entomol. 2002, 41, 375–377. [Google Scholar] [CrossRef]
- Mora, P.; Vela, J.; Ruiz-Mena, A.; Palomeque, T.; Lorite, P. Characterization and transcriptional analysis of a subtelomeric satellite DNA family in the ladybird beetle Henosepilachna argus (Coleoptera: Coccinellidae). Eur. J. Entomol. 2017, 114, 481–487. [Google Scholar] [CrossRef][Green Version]
- Mora, P.; Vela, J.; Ruiz-Mena, A.; Palomeque, T.; Lorite, P. Isolation of a pericentromeric satellite DNA family in Chnootriba argus (Henosepilachna argus) with an unusual short repeat unit (TTAAAA) for beetles. Insects 2019, 10, 306. [Google Scholar] [CrossRef]
- Gregory, T.R.; Nicol, J.A.; Tamm, H.; Kullman, B.; Kullman, K.; Leitch, I.J.; Murray, B.G.; Kapraun, D.F.; Greilhuber, J.; Bennett, M.D. Eukaryotic genome size databases. Nucleic Acids Res. 2007, 35, D332–D338. [Google Scholar] [CrossRef] [PubMed]
- Lorite, P.; Chica, E.; Palomeque, T. G-banding and chromosome condensation in the ant, Tapinoma nigerrimum. Chromosome Res. 1996, 4, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Sumner, A.T. A simple technique for demonstrating centromeric heterochromatin. Exp. Cell Res. 1972, 75, 304–306. [Google Scholar] [CrossRef]
- Palomeque, T.; Muñoz-López, M.; Carrillo, J.A.; Lorite, P. Characterization and evolutionary dynamics of a complex family of satellite DNA in the leaf beetle Chrysolina carnifex (Coleoptera, Chrysomelidae). Chromosome Res. 2005, 13, 795–807. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef]
- Altschul, S.F.; Stephen, F.; Thomas, L.; Madden, L.; Schaffer, A.A.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 4 June 2020).
- Rozek, M.; Holecová, M. Chromosome numbers, C-banding patterns and sperm of some ladybird species from Central Europe (Coleoptera, Coccinellidae). Folia Biol. 2002, 50, 17–21. [Google Scholar]
- Smith, S.G. Extreme chromosomal polymorphism in a coccinellid beetle. Experientia 1956, 12, 52–55. [Google Scholar] [CrossRef]
- Lyapunova, E.A.; Vorontsov, N.N.; Yadav, J.S.; Korablev, V.P.; Yanina, Y.; Gokhman, V.E. Karyological investigations on seven species of coccinellid fauna of USSR (Polyphaga: Coleoptera). Zool. Anz. Jena 1984, 212, 185–192. [Google Scholar]
- Blackmon, H.; Demuth, J.P. Coleoptera Karyotype Database. Coleopterists. Bull. 2015, 69, 174–175. [Google Scholar] [CrossRef]
- International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010, 8, e1000313. [Google Scholar] [CrossRef]
- Gregory, T.R.; Nedved, O.; Adamowicz, S.J. C-value estimates for 31 species of ladybird beetles (Coleoptera: Coccinellidae). Hereditas 2003, 139, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Gimenez, O.M.; Milani, D.; Lemos, B.; Castillo, E.R.; Martí, D.A.; Ramos, E.; Martins, C.; Cabral de Mello, D.C. Uncovering the evolutionary history of neo-XY sex chromosomes in the grasshopper Ronderosia bergii (Orthoptera, Melanoplinae) through satellite DNA analysis. BMC Evol. Biol. 2018, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruano, F.J.; Cabrero, J.; López-León, M.D.; Camacho, J.P.M. Satellite DNA content illuminates the ancestry of a supernumerary (B) chromosome. Chromosoma 2017, 126, 487–500. [Google Scholar] [CrossRef]
- Utsunomia, R.; Silva, D.; Ruiz-Ruano, F.J.; Goes, C.; Melo, S.; Ramos, L.P.; Oliveira, C.; Porto-Foresti, F.; Foresti, F.; Hashimoto, D.T. Satellitome landscape analysis of Megaleporinus macrocephalus (Teleostei, Anostomidae) reveals intense accumulation of satellite sequences on the heteromorphic sex chromosome. Sci. Rep. 2019, 9, 5856. [Google Scholar] [CrossRef]
- Ruiz-Ruano, F.J.; Navarro-Domínguez, B.; Camacho, J.P.M.; Garrido-Ramos, M.A. Characterization of the satellitome in lower vascular plants: The case of the endangered fern Vandenboschia speciosa. Ann. Bot. 2018, 123, 587–599. [Google Scholar] [CrossRef]
- Milani, D.; Bardella, V.B.; Ferretti, A.B.S.M.; Palacios-Gimenez, O.M.; Melo, A.S.; Moura, R.C.; Loreto, V.; Song, H.; Cabral-de-Mello, D.C. Satellite DNAs unveil clues about the ancestry and composition of B chromosomes in three grasshopper species. Genes 2018, 9, 523. [Google Scholar] [CrossRef]
- Lorite, P.; Carrillo, J.A.; Aguilar, J.A.; Palomeque, T. Isolation and characterization of two families of satellite DNA with repetitive units of 135 bp and 2.5 kb in the ant Monomorium subopacum (Hymenoptera, Formicidae). Cytogen. Genome Res. 2004, 105, 83–92. [Google Scholar] [CrossRef]
- Lorite, P.; Carrillo, J.A.; Tinaut, A.; Palomeque, T. Evolutionary dynamics of satellite DNA in species of the genus Formica (Hymenoptera, Formicidae). Gene 2004, 332, 159–168. [Google Scholar] [CrossRef]
- Ando, T.; Matsuda, T.; Goto, K.; Hara, K.; Ito, A.; Hirata, J.; Yatomi, J.; Kajitani, R.; Okuno, M.; Yamaguchi, K.; et al. Repeated inversions within a pannier intron drive diversification of intraspecific colour patterns of ladybird beetles. Nat. Commun. 2018, 9, 3843. [Google Scholar] [CrossRef] [PubMed]
- Magro, A.; Lecompte, E.; Magne, F.; Hemptinne, J.L.; Crouau-Roy, B. Phylogeny of ladybirds (Coleoptera: Coccinellidae): Are the subfamilies monophyletic. Mol. Phylogenet. Evol. 2010, 54, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Escalona, H.E.; Zwick, A.; Li, H.; Li, J.; Wang, X.; Pang, H.; Hartley, D.; Jermiin, L.S.; Nedvěd, O.; Misof, B.; et al. Molecular phylogeny reveals food plasticity in the evolution of true ladybird beetles (Coleoptera: Coccinellidae: Coccinellini). BMC Evol. Biol. 2017, 17, 151. [Google Scholar] [CrossRef] [PubMed]
- Pezer, Z.; Brajković, J.; Feliciello, I.; Ugarković, D. Satellite DNA-mediated effects on genome regulation. Genome Dyn. 2012, 7, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Bierhoff, H.; Postepska-Igielska, A.; Grummt, I. Noisy silence: Non-coding RNA and heterochromatin formation at repetitive elements. Epigenetics 2013, 9, 1–8. [Google Scholar] [CrossRef]
- Plohl, M.; Meštrović, N.; Mravinac, B. Centromere identity from the DNA point of view. Chromosoma 2014, 123, 313–325. [Google Scholar] [CrossRef]
- Pavlek, M.; Gelfand, Y.; Plohl, M.; Meštrović, N. Genome wide analysis of tandem repeats in Tribolium castaneum genome reveals abundant and highly dynamic tandem repeat families with satellite DNA features in euchromatic chromosomal arms. DNA Res. 2015, 22, 387–401. [Google Scholar] [CrossRef]
- Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-Carvalho, M.; Chaves, R. Decoding the role of satellite DNA in genome architecture and plasticity-an evolutionary and clinical affair. Genes 2020, 11, 72. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SatDNA Family | Primer Name | Primer Sequence |
|---|---|---|
| HvarSat01-277 | Hvar-CL1-F | 5′ ACTCTCTATCCCTACCCG |
| Hvar-CL1-R | 5′ AAATCAGTTGAGCCTGAG | |
| HvarSat02-127 | Hvar-CL3-F | 5′ AAAAATCGAGAGTTTTCG |
| Hvar-CL3-R | 5′ TTCATCTCATTCTGACGC | |
| HvarSat03-217 | Hvar-CL13-F | 5′ CTGGCAAATGCAAGACTTC |
| Hvar-CL13-R | 5′ TACGATGTATTCACGACG | |
| HvarSat04-487 | Hvar-CL12-F | 5′ GAAGATCTGATCCCACTG |
| Hvar-CL12-R | 5′ TCAGAGGCAATCTAGGAG |
| Name | Genome Proportion | Repeat Unit Length (bp) | A+T Percentage | Kimura Divergence (%) |
|---|---|---|---|---|
| HvarSat01-277 | 9.37% | 277 | 65.3% | 5.71% |
| HvarSat02-127 | 2.04% | 127 | 65.4% | 4.46% |
| HvarSat03-217 | 0.92% | 217 | 59.9% | 6.67% |
| HvarSat04-487 | 0.62% | 487 | 64.1% | 3.25% |
| HvarSat05-324 | 0.56% | 324 | 59.9% | 5.78% |
| HvarSat06-175 | 0.49% | 175 | 65.1% | 4.91% |
| HvarSat07-2000 | 0.13% | 2000 | 68.2% | 1.31% |
| HvarSat08-972 | 0.10% | 972 | 66.8% | 2.38% |
| HvarSat09-292 | 0.09% | 292 | 59.9% | 3.73% |
| HvarSat10-91 | 0.07% | 91 | 63.8% | 9.35% |
| HvarSat11-141 | 0.07% | 141 | 63.9% | 4.89% |
| HvarSat12-150 | 0.05% | 150 | 52.7% | 1.19% |
| HvarSat13-148 | 0.04% | 148 | 58.1% | 6.68% |
| HvarSat14-309 | 0.03% | 309 | 70.3% | 21.34% |
| HvarSat15-158 | 0.03% | 158 | 63.2% | 8.45% |
| HvarSat16-87 | 0.03% | 87 | 74.7% | 25.74% |
| HvarSat17-176 | 0.024% | 176 | 65.2% | 10.22% |
| HvarSat18-191 | 0.020% | 191 | 49.2% | 4.44% |
| HvarSat19-143 | 0.018% | 143 | 64.4% | 5.79% |
| HvarSat20-141 | 0.016% | 141 | 60.3% | 7.51% |
| HvarSat21-152 | 0.015% | 152 | 62.5% | 8.63% |
| HvarSat22-145 | 0.011% | 145 | 63.5% | 9.27% |
| HvarSat23-378 | 0.011% | 378 | 60.4% | 2.28% |
| HvarSat24-105 | 0.008% | 105 | 53.3% | 2.98% |
| HvarSat25-150 | 0.007% | 150 | 69.3% | 8.2% |
| HvarSat26-164 | 0.006% | 164 | 66.4% | 5.11% |
| HvarSat27-41 | 0.004% | 41 | 68.3% | 9.04% |
| HvarSat28-57 | 0.003% | 57 | 45.6% | 6.86% |
| HvarSat29-169 | 0.003% | 169 | 69.2% | 5.5% |
| Telomeric | 0.17% | 5 | 60.0% | 0.23% |
| Total | 14.93% | |||
| Mean | 265.73 | 62.63% | 6.73% | |
| SD | 372.02 | 6.27% | 5.30% | |
| Median | 155.00 | 63.85% | 5.75% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mora, P.; Vela, J.; Ruiz-Ruano, F.J.; Ruiz-Mena, A.; Montiel, E.E.; Palomeque, T.; Lorite, P. Satellitome Analysis in the Ladybird Beetle Hippodamia variegata (Coleoptera, Coccinellidae). Genes 2020, 11, 783. https://doi.org/10.3390/genes11070783
Mora P, Vela J, Ruiz-Ruano FJ, Ruiz-Mena A, Montiel EE, Palomeque T, Lorite P. Satellitome Analysis in the Ladybird Beetle Hippodamia variegata (Coleoptera, Coccinellidae). Genes. 2020; 11(7):783. https://doi.org/10.3390/genes11070783
Chicago/Turabian StyleMora, Pablo, Jesús Vela, Francisco J. Ruiz-Ruano, Areli Ruiz-Mena, Eugenia E. Montiel, Teresa Palomeque, and Pedro Lorite. 2020. "Satellitome Analysis in the Ladybird Beetle Hippodamia variegata (Coleoptera, Coccinellidae)" Genes 11, no. 7: 783. https://doi.org/10.3390/genes11070783
APA StyleMora, P., Vela, J., Ruiz-Ruano, F. J., Ruiz-Mena, A., Montiel, E. E., Palomeque, T., & Lorite, P. (2020). Satellitome Analysis in the Ladybird Beetle Hippodamia variegata (Coleoptera, Coccinellidae). Genes, 11(7), 783. https://doi.org/10.3390/genes11070783