1. Introduction
Grapes are not only tasty, but also one of the most economically and culturally important crops. They are used for both winemaking and fresh (‘table’) consumption. Grape is unique, not only because it is a major global perennial crop, but also because of its historical and cultural connections with the development of humans. According to the OIV (International Organization of Vine and Wine) the surface area of the world vineyard is estimated at 7.4 million hectares, with a production of 76 million tons of fresh grapes and 260 million hectoliters of wine (
http://www.oiv.int/). Grapevine (
Vitis Vinifera) is one of the oldest of the cultivated plants for which living progenitors still exist. The broad geographic area of the wild grapes and a bewildering number of different forms expanding from Europe to Asia and Caucasus continue to intrigued researchers, with a crucial question about the origin and domestication of the grape. Archaeological and historical studies suggested that the cultivation of the domesticated grape (
V. vinifera L. subsp. sativa) started about 10,000 to 8000 years ago, from its putative wild ancestor (
V. vinifera L. subsp. sylvestris), and the primary center of domestication was located between the Near East [
1] and the Transcaucasian region [
2]. These regions were populated by the Neolithic Shulaveri-Shomu, and later Kura-Araxes cultures, in the South Caucasus and Fertile Crescent regions, ranging from (today’s) Georgia, through Azerbaijan, Armenia, northern Iran and eastern Anatolia [
3]. Later, grapevine disseminated into the Southern Balkans and East Mediterranean Basin, then to the Western Europe and, finally, domesticated grapes were introduced to Central Europe during the first millennium BCE [
4]. Hereby, wine grapes have followed human civilization from South Caucasus, southwards and westwards, spread first by seafaring Phoenicians and Greeks, and later affected by Roman and Ottoman Empires throughout the Mediterranean world [
5]. During its spreading across the regions, grapes slowly mutated and adapted to their environments. The slow divergence over thousands of years in combination with the spontaneous hybridization, somatic variation and selection by humans created the incredible diversity of the more than 6000 cultivated varieties, which, in contrast to its wild progenitor, is more diverse and heterozygous according to OIV (
http://www.oiv.int/). Different authors evidenced the presence of secondary domestication centers, where spontaneous hybridizations among wild plants and cultivated forms or targeted selection, created the pattern of the modern Western European cultivars [
4]. M.A. Negrul, an outstanding pioneer researcher of vine, subdivided the varieties based on geographic origin and morpho-ecological traits into the ecotypes occidentalis (France, Germany, Spain, Portugal), pontica (Asia Minor, Romania, Hungary, Greece, Georgia, Bessarabia) and orientalis (Armenia, Iran, Afghanistan, Azerbaijan and Central Asia), where the primary center of a given plant is the region of the richest genetic diversity [
6].
In the last years, whole-genome studies of vine cultivars using genotyping and sequencing technologies have added novel knowledge about the diversity, geographical relatedness, historical origin, phenotype associations, genetic markers and distribution paths of the vine [
4,
7,
8,
9,
10,
11]. Genetic relatedness between cultivated (
V. Vinifera L. subsp. sativa) and wild (
V. Vinifera L. subsp. sylvestris) grapes suggest at minimum two separate domestication events, one derived from the Transcaucasian wild grape and another one in Western Europe, where cultivars experienced introgression from local wild grape [
4,
7]. Associations between candidate genes and important agronomic traits, such as berry shape and aromatic compounds, provide possible genetic targets for grapevine improvement [
9]. Breeding for larger berries has been related to genetic signatures of positive selection for table grapes in geographic regions, where alcohol was prohibited by religious rules [
8]. The further impact of genomic studies will depend on the understanding of the genotype-phenotype associations, especially for complex traits governed by polygenic architecture, genotype-environment interactions in different geographic regions [
11]. These tasks challenge activities and research infrastructure for collecting cultivar accessions, sequencing and data deposition, for providing phenotype-genotype databases and vine passporting projects, and, last but not least, the development of bioinformatics methods and tools which enable an analysis of these data, in terms of knowledge mining and feature extraction, in an easy and intuitive fashion.
Here, we apply SOM (self-organizing maps) portrayal [
12], a neural network-based machine learning method with strong visualization capabilities to genome-wide Single nucleotide polymorphism (SNP) data of nearly eight hundred grapevine cultivars collected from Middle Asia in the East to Iberian peninsula in the West and from overseas regions [
11]. SOM portrayal has been developed by us for the detailed analysis of high-dimensional omics data, including diversity and developmental issues, feature selection, knowledge mining and phenotype association of transcriptomic [
13,
14,
15], epigenetic [
16,
17], proteomics [
18] and genetic data [
19], and of combinations of them [
20]. In the context of plants, SOM-portrayal has been previously applied by us for the typing of algae of the genus Prototheca [
21] and by others for studying early seed maturation in garden pea [
22]. This work aims at providing a prototypic application of SOM machine learning and linked downstream analytics to illustrate its potency for studying plant genomes.
Strengths of the applied method are dimension reduction and visualization capabilities [
23]. Particularly, our method generates accession-specific images of the data landscapes. These personalized omics-portraits provide options for the intuitive evaluation of feature space, and for mutual comparisons between the individual accessions. The recent application of SOM-portrayal to population-level distributions of disease-related human SNP-variants demonstrated its capability to extract genetic features and to describe genetic diversity, based on the topology of the SNP-landscapes [
19].
This study aims at applying SOM-portrayal to the genetics of vine, in order to generate SNP-landscape of grape genotypes, to relate its topology to the geographical distribution, to possible paths of cultivar distribution, to selected genetic markers, and link them to grape utilization. In gastronomy, the sommelier is the wine ‘waiter’ (or ‘waitress’), who recommends combining wine with food based on traditional knowledge about the character of wine varieties, their taste and geographic origin, and thus to support the decision-making of guests for a successful dinner. In analogy, our ‘SOMmelier’-approach aims at supporting understanding genetic diversity and relatedness of grapevine genomes in the context of their geographic and historical background by application of SOM portrayal.
2. Materials and Methods
2.1. Data and Preprocessing
Grape cultivars data: grapevine genetic SNP- and phenotype data were taken from [
11]. The data set consists of 783 grapevine samples originated from 4 grapevine collections (
Table S1, Passport data of 783 cultivars included in the study) collected from eleven geographic regions, ranging from Middle Asia to Iberia and New World accessions. The genotype data matrix of 10,207 SNPs for 783 unique samples was taken from
https://urgi.versailles.inra.fr/Species/Vitis/Data-Sequences/Genotyping-data repository.
Human population data: For a comparison of genomic relatedness between worldwide geographic strata, we analyzed genome-wide microarray SNP data (Illumina 650Y arrays) from the Human Genome Diversity Project (HGDP) [
24], as described previously [
19]. This data consists of 650,0000 SNPs for 940 individuals from 8 geographical regions (Africa, Europe, Middle East, South and Central Asia, East Asia, Oceania, and America). Genotypes for 99 individuals of Armenian ethnicity (Illumina Human Omniexpress microarray platform) were taken from a publication by Haber et al. [
25]
2.2. Conceptual Overview
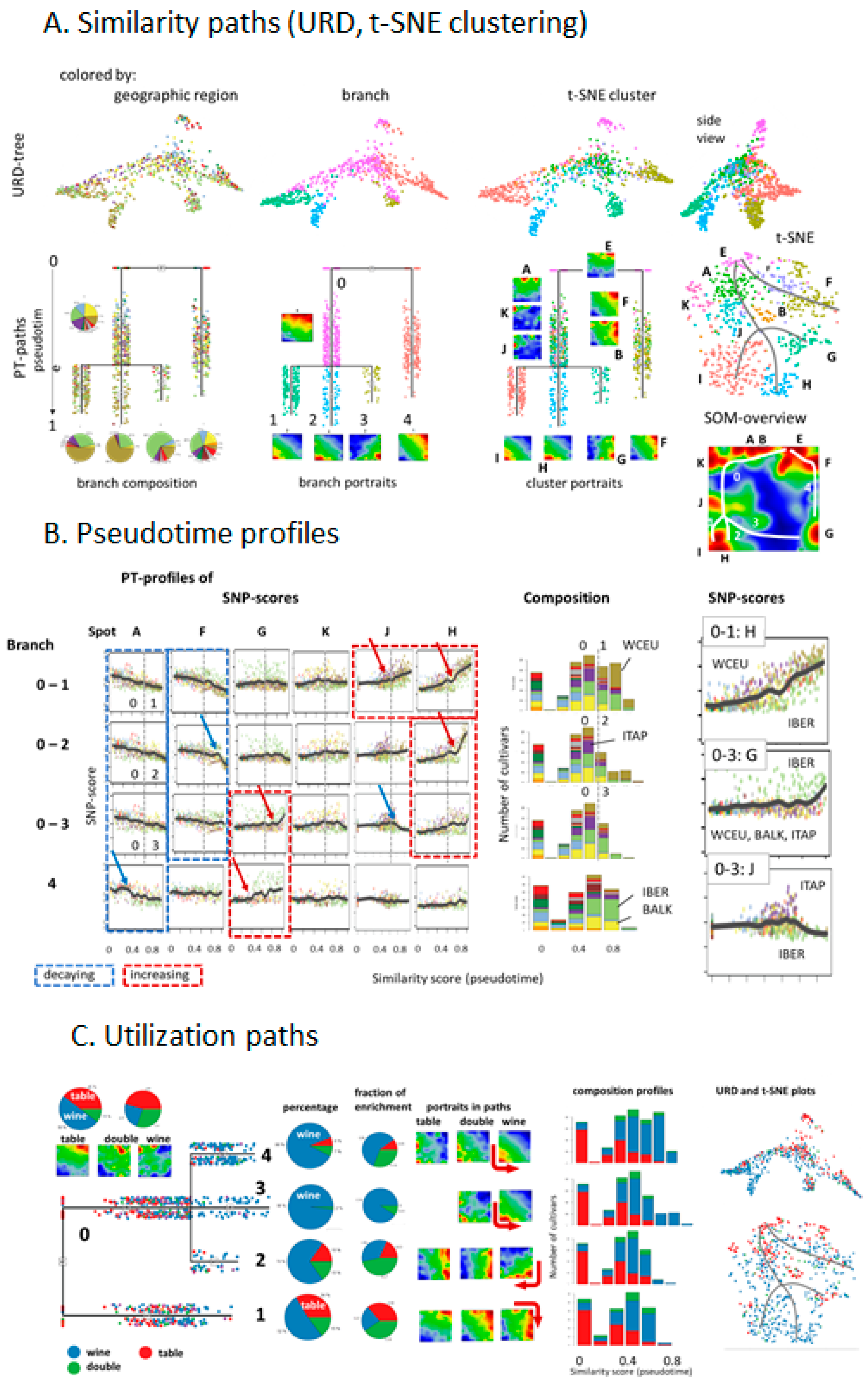
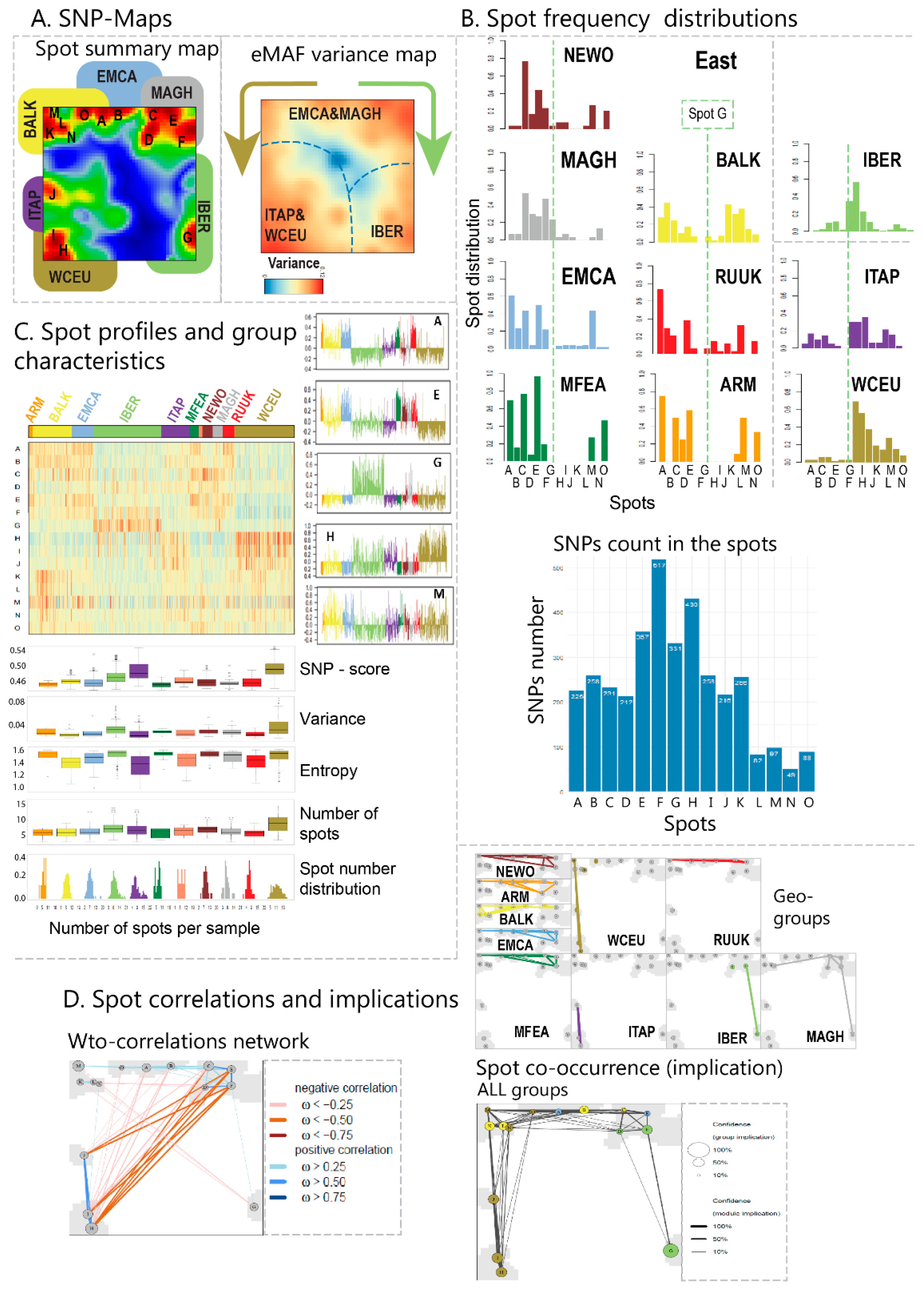
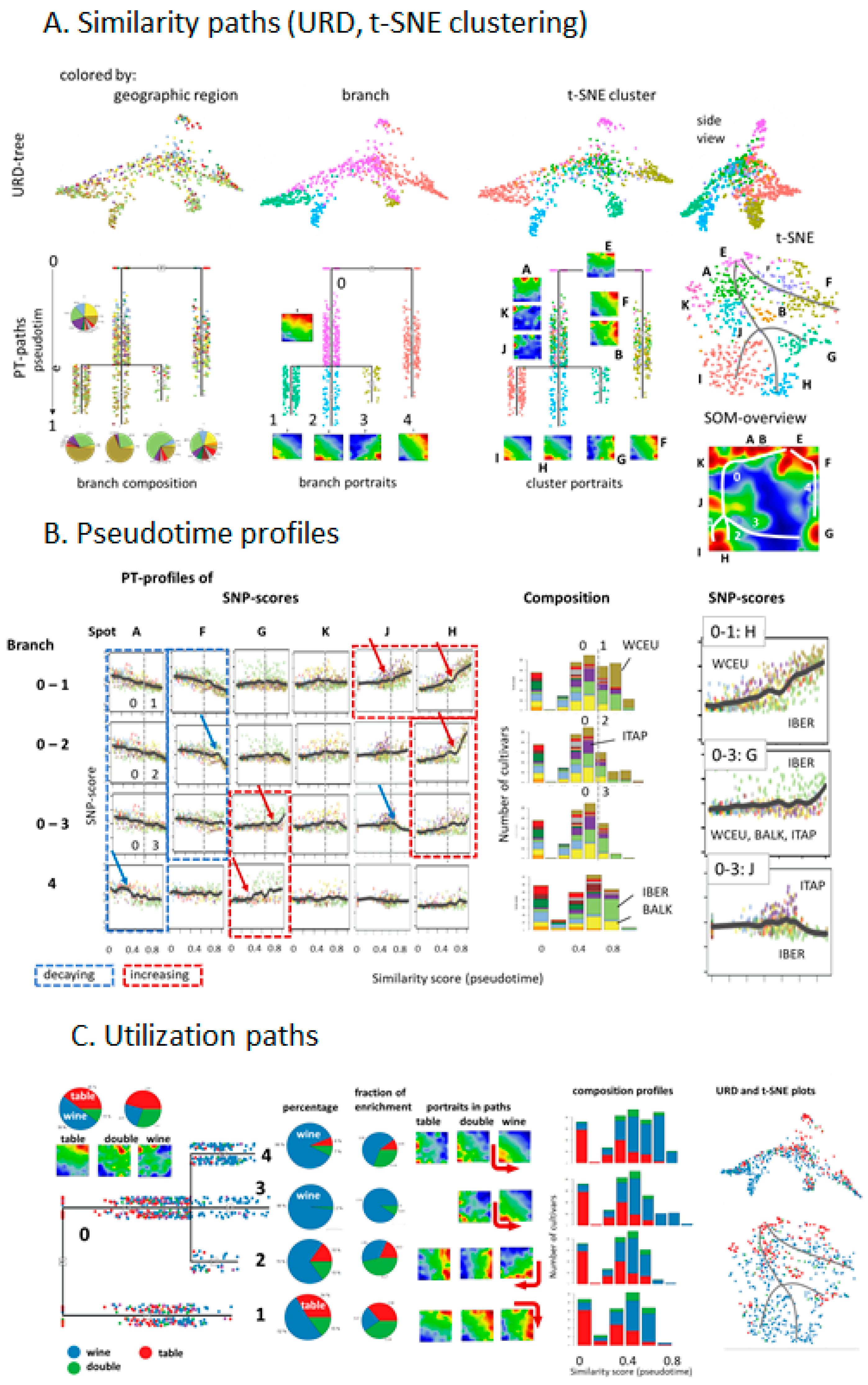
Our analysis concept is based on the following main ingredients: (i) transformation of SNP data into excess minor allele frequencies (eMAF), which increases sensitivity for subtle differences between the genomes of the vine accessions; (ii) dimension reduction of the more than 10,000 SNP data into a matrix of 2500 so-called ‘meta-SNPs’, using unsupervised clustering and their visualization in terms of individual accession portraits using SOM machine learning. (iii) further reduction of dimension by segmentation of the SNP-portraits into about one-to-two dozen so-called spot clusters of correlated SNPs, which serve as fingerprint features of the vine genomes; (iv) diversity analysis of accessions using different approaches (principal component analysis, correlation networks, t-SNE, minimum spanning tree). Each of the approaches used enables discovering different aspects of the mutual relatedness between the cultivars. (v) So-called pseudotime analysis as a special type of diversity analysis to analyze genetic similarities in terms of multibranched trees with possible impact for temporal diversification of vine cultivars; (vi) phenotype associations to map information of vine utilization on the SNP landscapes.
2.3. Allele Coding and SNP-Score
In order to further process the data, we coded the genotype of each SNP using a trinary code, with ‘0′ for homozygous major alleles (AA), ‘1′ for heterozygous alleles (Aa and aA), and ‘2′ for homozygous minor alleles (aa).
First, let us consider one SNP in the data set of N cases, e.g., of N =783 cultivar samples. The fractions of the three genotypes of this SNP are defined as pij = Nij/N (i,j = A, a), where Nij is the number of SNPs with the respective allele in the data set. The minor allele frequency of a SNP is MAF = 2 paa + 1 (paA + pAa) with paa < pAA. With the SNP-code introduced above one finds for its mean value averaged over all cases, <SNP-code> = pAA * 0 + (pAa + paA) * 1 + paa * 2 = 2 MAF, i.e., the mean SNP-code of a SNP equals twice its MAF. For further data processing, we define a SNP-score by centralizing the SNP-code of each SNP with respect to its mean value, SNP-score = SNP-code – 2 MAF. One obtains SNP-score = −2 MAF for the major allelic SNPs, SNP-score = 1 – 2 MAF for heterozygous SNPs and SNP-score = 2 (1 – MAF) for the minor allelic SNPs.
Second, let us consider a group of n correlated SNPs in the data set, and calculate the mean of their SNP-score in one selected sample. In the next subsection, we show that such a group of SNPs is given, e.g., by a meta-SNP, or a ‘spot-module’ as obtained below in SOM-analysis. Analogous consideration as above deliver the result for the group-averaged SNP-score, <SNP-score> = 2 (MAF - <MAF>) where <…> here denotes group averaging referring to one selected sample. Hence, the group-averaged SNP-score estimates the deviation of the mean MAF of the considered SNPs, in a certain sample from their mean in the considered population. Accordingly, it can be understood as an excess MAF-value (eMAF), where positive SNP-scores mean higher frequencies than in the population while negative values refer to reduced frequencies. The centralization of each feature (here the SNP-code of each SNP) with respect to its mean value averaged over all samples is applied as a standard preprocessing step in SOM analysis. Features space of centralized values is more sensitive to subtle differences between samples than the feature space of non-centralized values.
2.4. SOM Portrayal and Spot Detection
SOM-portrayal of SNP data was performed as described previously [
19]. In short: our SOM implementation used a ternary code with the values 0, 1 and 2 for major homozygous, heterozygous and minor heterozygous genotypes, respectively, as introduced above. Next, SNP data were mean centralized and then clustered using the self-organizing map (SOM) machine learning. SOM training translates the original data matrix consisting of the allele scores of N = 10,206 SNPs collected from M = 783 cultivar accessions into a data matrix of reduced dimensionality of K = 2500, so-called meta-SNP profiles. Hereby, the term ‘profile’ denotes the vector of eMAF score values across the cultivars. The SOM training algorithm distributes the SNPs over the K micro-clusters of meta-SNPs, by minimizing the
Euclidean distance between the SNP- profiles as a similarity measure. It ensures that SNPs with similar profiles cluster together in the same or in closely located meta-SNPs. Each meta-SNP profile can be interpreted as the mean profile averaged over all SNP profiles of the respective meta-SNP cluster. For each cultivar accession, the meta-SNP score obtained provides the excess MAF (eMAF, see above). It is defined as the difference between the mean minor allele frequency (MAF of meta-SNP) of all SNPs collected in this meta-SNP in this particular sample, minus the mean MAF of these SNPs averaged over all samples. The eMAF values of each cultivar accession are visualized by arranging them into a two-dimensional M = 50 × 50 grid, and by using a red to blue color-code for maximum to minimum eMAF-values in each of the grid images. These images ‘portray’ the genetic landscape of each accession studied in units of the eMAF SNP-score. We used SOM as implemented in the “oposSOM” R package [
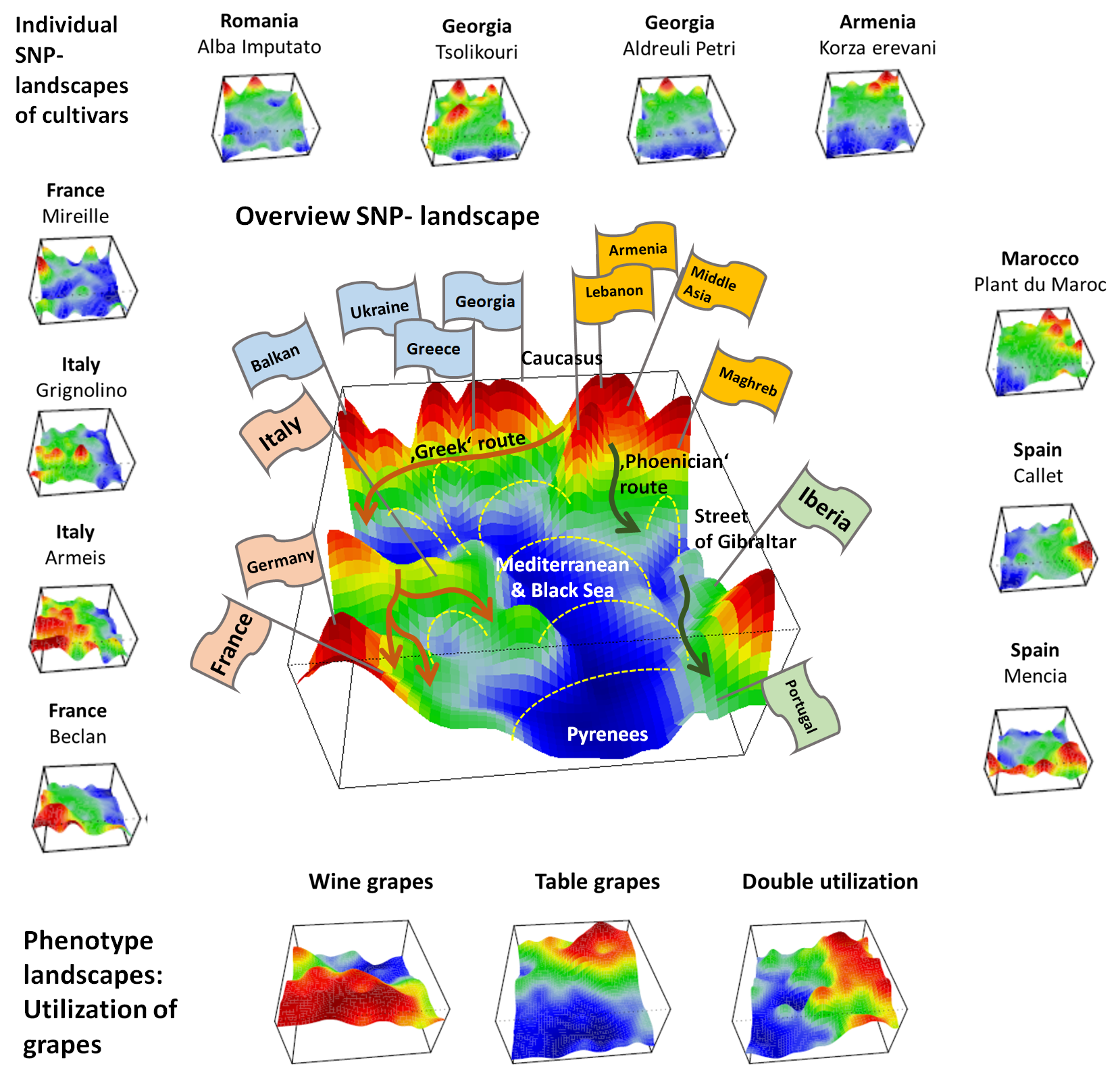
23]. Cultivars were labelled according to the geographical region where they were selected (see [
4] for details) and, alternatively, using different cluster assignments (see below). Mean SNP-SOM portraits of cultivars from the same geographic regions were obtained by averaging the meta-SNP values of the respective individual SNP-portraits. The self-organizing properties of the SOM algorithm generates red spot-like regions referring to correlated SNP-profiles showing high eMAF-values in the respective accession portraits. We used segmentation algorithms developed previously [
23] to extract so-called spot-clusters from these (red) regions. Each of these spot-clusters includes hundreds of SNPs.
2.5. Phenotype Association, Accession Diversity, Pseudotime Analysis and Clustering
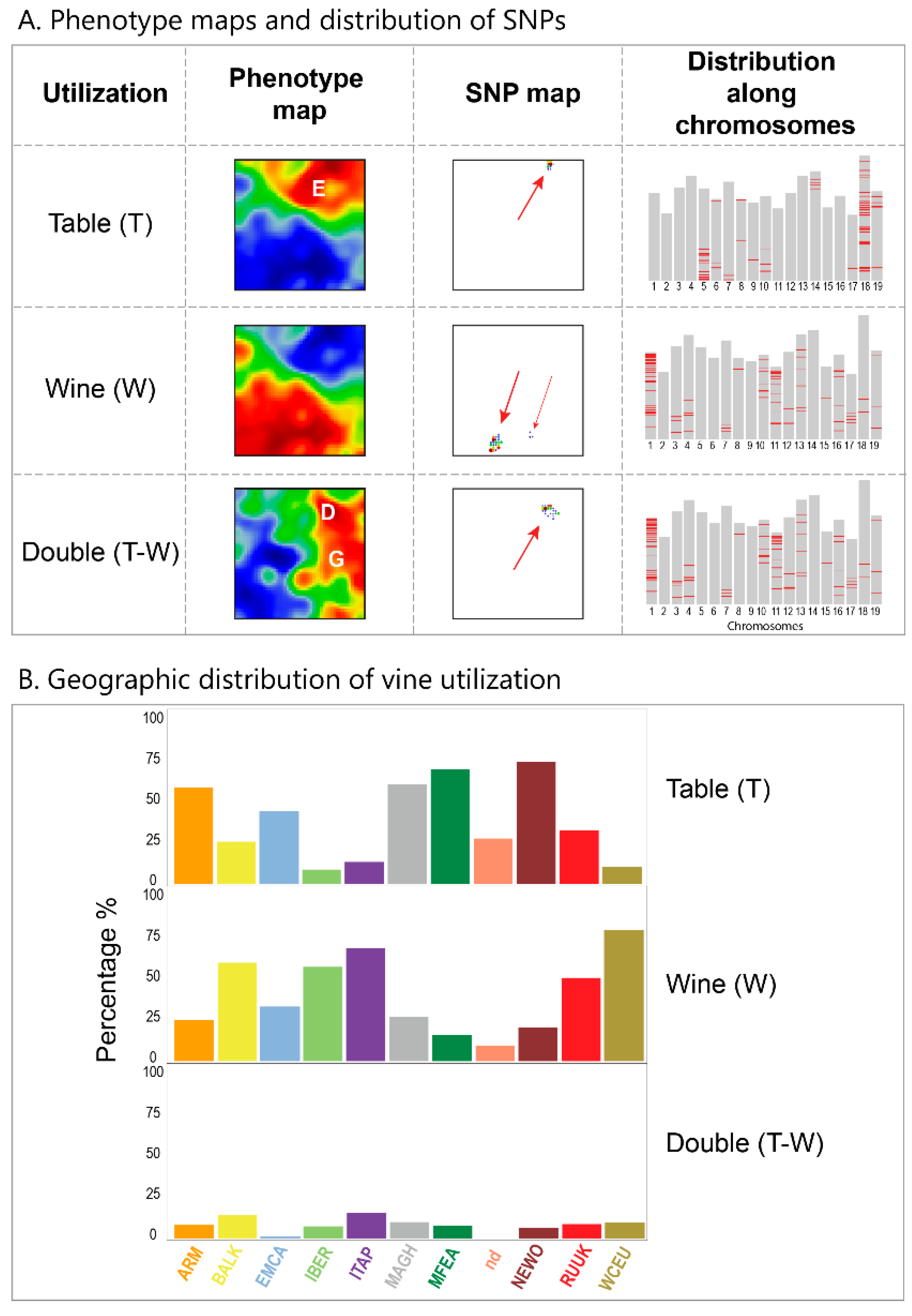
Categorical phenotypes such as the utilization of grapes for table or wine usage were associated with the SOM SNP-landscape by performing ANOVA (analysis of variance) of SNP-metagenes of the sub-collection of cultivars of a certain phenotype (e.g., of all cultivars of table-vine usage) and coloring the meta-SNP pixels in the SOM according to the obtained p-value (in units of -log(p)), from red (high) to blue (low). Phenotype correlation maps were obtained by calculating the point biserial correlation between the eMAF profile of each metagene and the respective phenotype profile, which is given, e.g., for ‘table vine utilization’ by categorical values ‘0′ (no table usage) and ‘1′ (table usage). The metagenes of the map were then colored between red (maximum correlation) and blue (minimum correlation). Point serial correlation de facto provides difference portraits between cultivars of the respective phenotype and all others, e.g., between the mean portrait of cultivars of wine utilization, minus the mean portraits of cultivars of non-wine utilization.
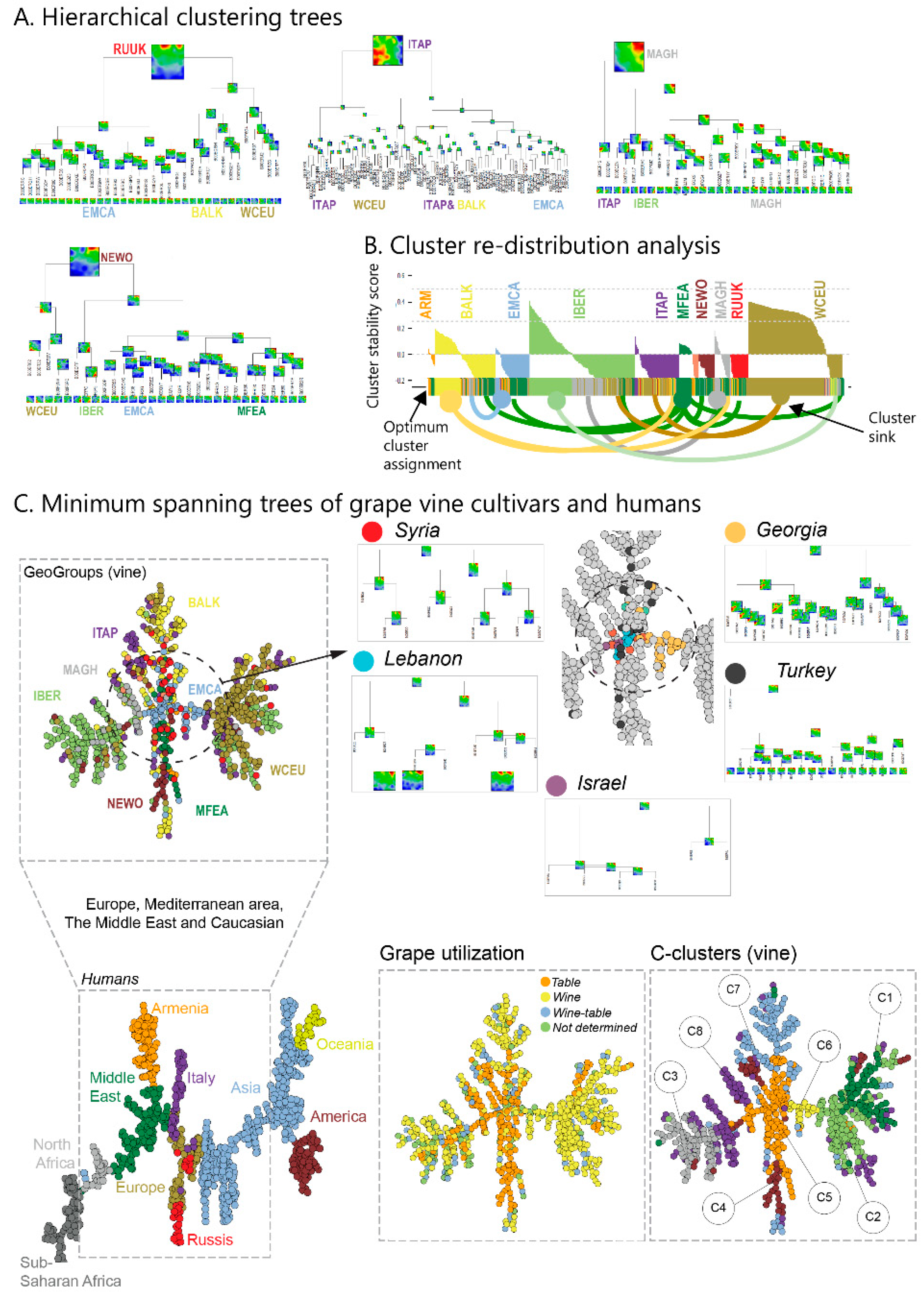
Accession diversity analysis was performed based on meta-SNPs using principal component analysis (PCA), similarity net and minimum spanning tree plots, based on correlation metrics between the individual SOM-portraits as implemented in oposSOM [
23]. t-SNE (t-distributed Stochastic Neighbour Embedding), URD-plots and pseudotime plots were generated using the program ‘URD’ [
26]. URD estimates multibranched developmental trajectories based on mutual similarities between the accessions, a k-nearest neighbor graph presentation and directed (from ‘root’ to ‘tip’) simulation of a diffusion-like process. It provides a pseudotime (PT) value between zero and unity for each accession, where values near zero mean closer similarity to the root accessions, and values near unity mean closer similarity to tip-accessions. URD generates branched tree structure by joining diffusion trajectories passing through the same accessions.
Grapevine cultivars were stratified according to the geographic region in agreement with [
10], where they were collected and using an eight cluster C1–C8 (C-clusters) division, according to [
11]. Independent ‘t-SNE’ clustering was performed using the Jaccard-algorithm implemented in ‘URD’ software [
26], which provided ten clusters (see below).
4. Discussion
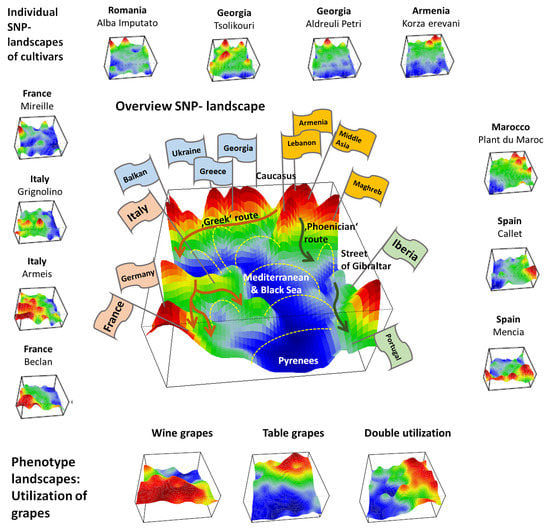
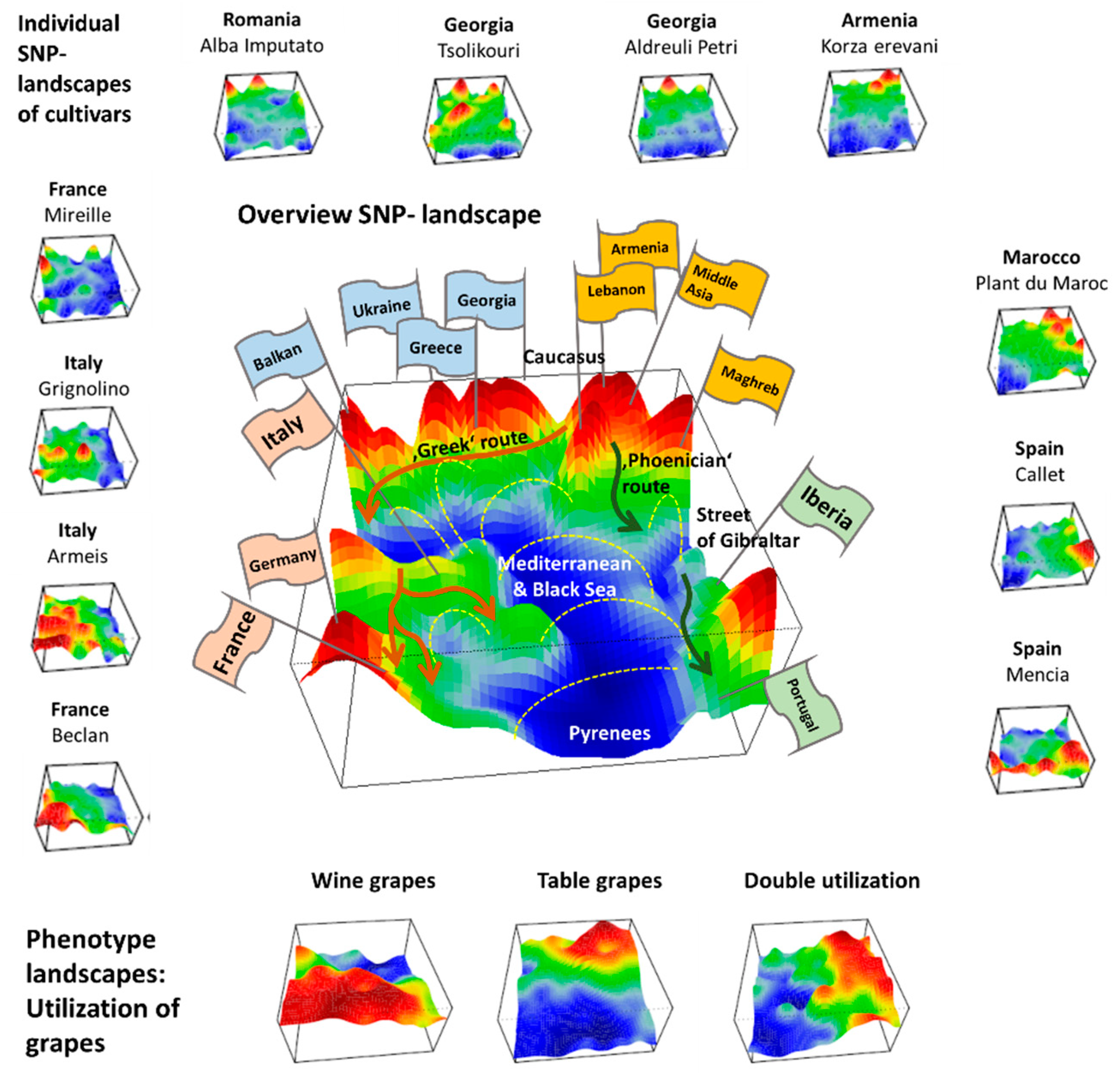
We applied SOM artificial neural network-based machine learning to genome-wide SNP data of 783 grapevine cultivars, in order to visualize and to analyze the landscape of grapevine genomes, in terms of topological features such as ‘mountains’ and ‘valleys’, referring to positive and negative eMAF values, respectively (
Figure 7).
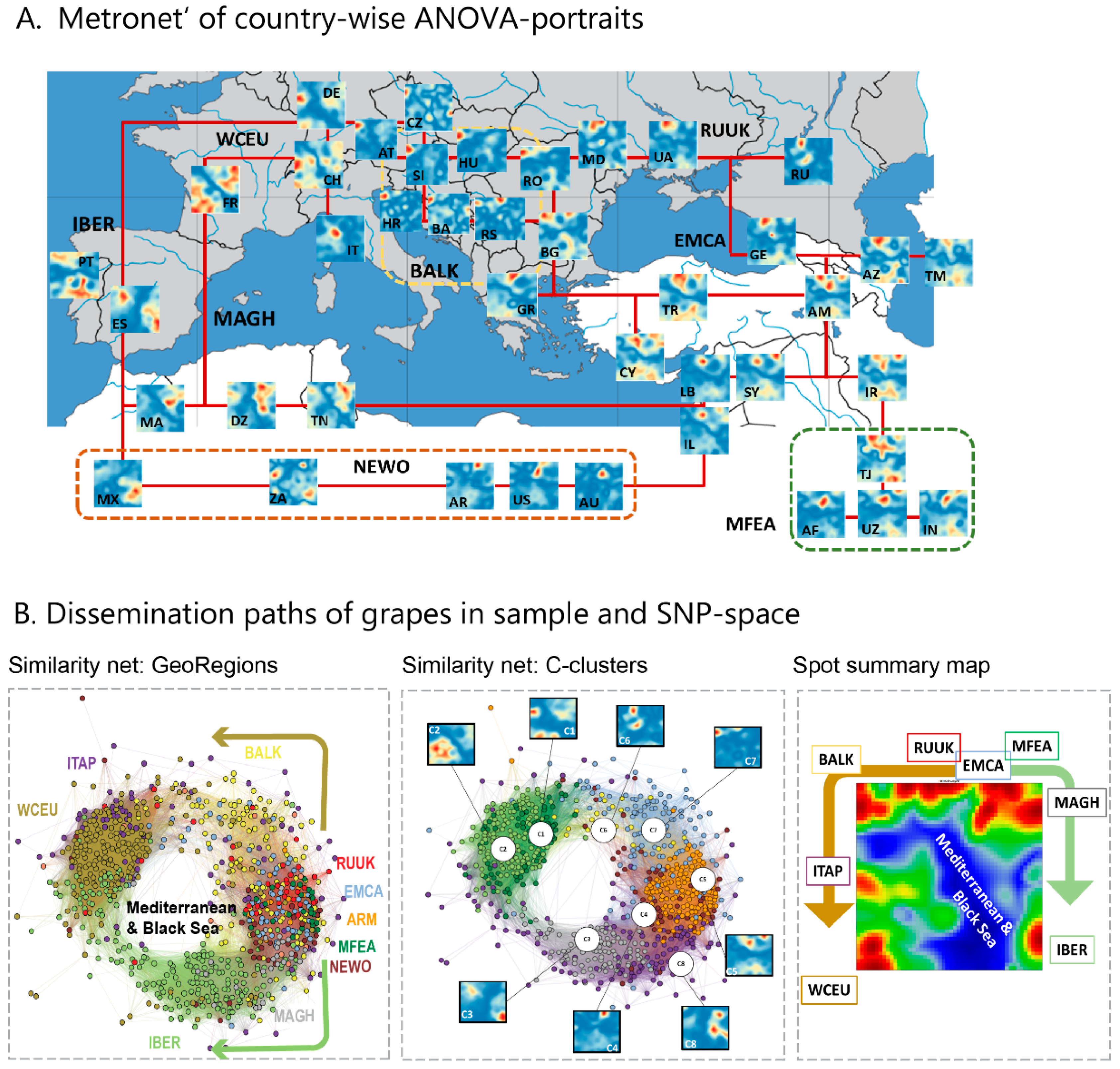
We complemented this genetic feature (SNP-) landscape by grapevine accession-similarity plots. Each of them visualizes different aspects of the mutual relatedness of cultivars. Principal component plots distribute accessions in (eMAF) distance-scale. It results in local overcrowding of cultivars with similar genomes, e.g., from Near East, Caucasus and Middle Asia while Western European and Iberian cultivars separate into virtually different data clouds of large variability between sample genomes. PCA requires separate projection-plots to cover the essential properties of similarity space. The (correlation) similarity net projects multidimensional sample space into two dimensions. It reveals major routes of grape distribution around the Mediterranean Sea and the genetic barrier between Iberian and Western European cultivars, evident also in the feature landscape. In contrast, the minimum spanning tree (MST) generates path-like structures. For worldwide human genomes, the MST reflects roughly the ‘out-of-Africa’ migration history, in terms of a virtually linear path starting from Sub-Saharan Africa and ending in East Asia, with side branches towards Europe, America and Oceania. Interestingly, the grapevine MST splits into four mean branches, enriching West European, Iberian, Balkan and Middle East grapes, and with cultivars from South Caucasus and Fertile Crescent (Syria, Lebanon, East Anatolia) at their central crossroad. A detailed comparison of individual (ANOVA-) portraits further supports this result, using the feature landscapes in a country-wise resolution.
Overall, our genomic landscape and the different sample similarity plots are consistent with the historical knowledge and previous genetic findings of grapevine domestication and distribution [
7,
10]. Accordingly, cultivated grapes occurred initially in South Caucasus (Georgia, Armenia) and Fertile Crescent (East Anatolia and North Lebanon and Syria) [
29,
30]. Indeed, grapes from this region form the crossroad in the MST, presumably due to footprints of initial cultivation in their genomes. Grapes then distributed towards the ‘classical’, Mediterranean world into the west direction and into East towards Iran and the Middle East (Tadjikistan, Uzbekistan), Afghanistan and India. The northern and southern ways into the West agree with the distribution of settlements of Greeks (the Black Sea, including Crimea, Anatolia, Southern Italy and Sicily, Southern France and Northern Spain along the Northern coast of Mediterranean Sea) and Phoenicians (Lebanon, Carthago/Tunis, Maghreb and South-West Spain), respectively. Genomes of vines from the Italian Peninsula are very diverse and reflect links to almost all parts of the Mediterranean area (BALK, EMCA, MAGH, IBER) and Western Europe, presumably due to intense cultural exchange in the Greek world, and later within the Roman empire [
31].
Hence, grape growing and winemaking in classical time disseminated from ‘Iberia-to-Iberia’, starting in or near the ancient kingdom Iberia in Middle Georgia in South Caucasus in the East and ending at the Iberian Peninsula in the west. The combined action of selection, breeding, admixture and migration have shaped the cultivated grape diversity. Substantial genetic diversity has been maintained, subsequent to domestication derived from Transcaucasian Wild Grape, possibly due to several events of introgression from local wild vine Silvestris-varieties, Wild grapes (
V. vinifera ssp. sylvestris), particularly in Western Europe [
7]. Although Mediterranean and Black Seas served rather as highways of cultural exchange than as barriers, vine distribution obviously followed primarily ‘country ways’ along with the coastal areas.
We visualize grape utilization in terms of phenotype maps which associate table, wine and double usage with different geographic regions (
Figure 7, part below). Grapes for fresh consumption (table vines) predominate in Eastern and North African areas, while wine utilization is found mostly in Western Europe (RUUK, BALK, WCEU, ITAP, IBER). This division has been associated with different religious rules concerning alcohol consumption in Islamic and Christian regions [
8]. Interestingly, the red region of wine-utilization covers the WCEU and IBER geographic regions, and thus it bridges the genetic border formed by the Pyrenees in the overview landscape (
Figure 7).
In order to extract additional information about the dissemination of cultivars, we applied pseudo-time (PT) analysis. This method has been originally developed for describing cell differentiation using single-cell transcriptomic data [
26,
32], and it was applied to study human cancer progression using single-cell and bulk transcriptome data [
14,
33]. In the context of this study, PT sorts and scales vine genomes according to their genetic similarity, into a directed, branched graph. Starting with the assumption of initial Eastern domestication of the grapevine PT then describes its dissemination in an East to West direction. Vines from the Caucasus and the East refer to small PT-values and vines from the Balkan and Italian peninsula mostly to intermediate PTs. Interestingly, the genetic characteristics of Western European and Iberian grapes follow different courses at later PTs along different branches, which eventually reflects different waves of cultivation, since the Roman Empire times. Particularly, recent pedigree analyses uncover a putative Middle Age cultivar melting pot, giving rise to many of today’s cultivars, suggesting even secondary domestication events taking place in Western Europe and the Iberian Peninsula ending in the cultivars that are used in viticulture today [
8]. Different PT-branches show different slopes and switching points in their grow regimes at later PTs. Table grapes mostly accumulate at early PTs. Hence, our ‘SOMelier’ transforms vine genomes into a landscape resembling the topology of the geographic regions of grape cultivation and/or the diversity of their genomes. Different methods of similarity analyses in SNP-feature and cultivar space enable extracting details of dissemination history and the utilization of grapes.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}