Displacement of Slow-Turnover DNA Glycosylases by Molecular Traffic on DNA

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Oligonucleotides and Enzymes
2.2. OGG1 Displacement From Its Complex with DNA
2.3. Decay of OGG1–DNA Covalent Intermediates
2.4. NEIL1 Displacement from Its Complex with DNA
2.5. Cas9 Displacement from Its Complex with DNA
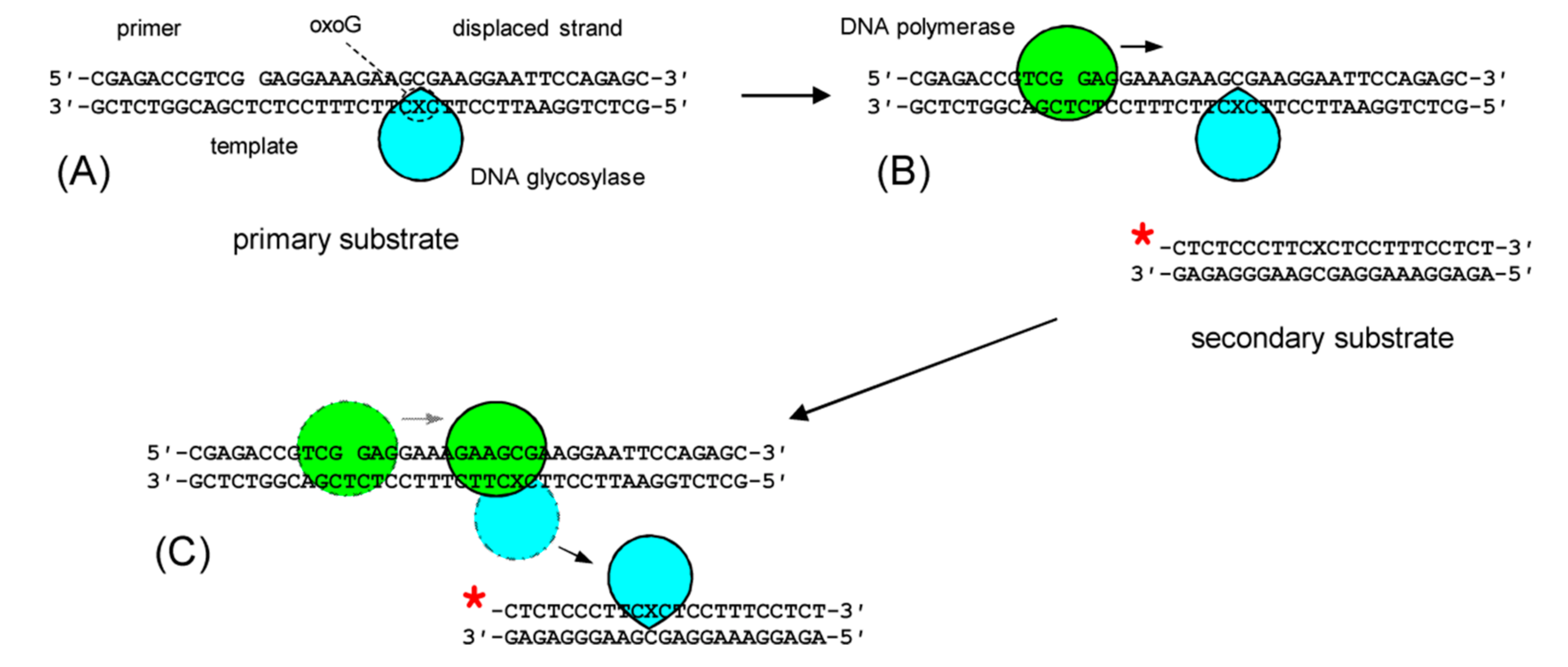
2.6. Primer Elongation in the Presence of Protein Obstacles
2.7. Microscale Thermophoresis
2.8. DNA-Guided Protein–Protein Docking
3. Results
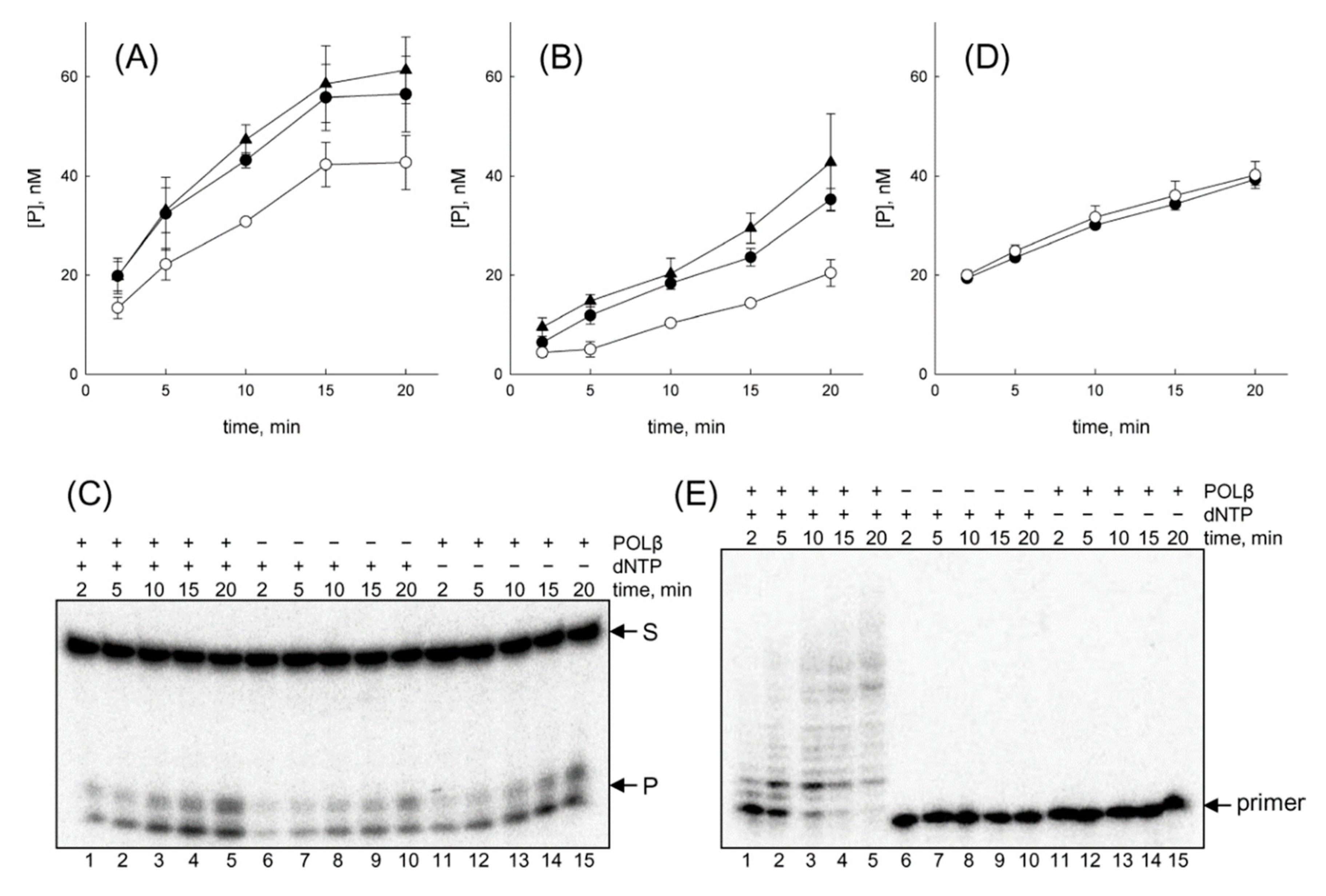
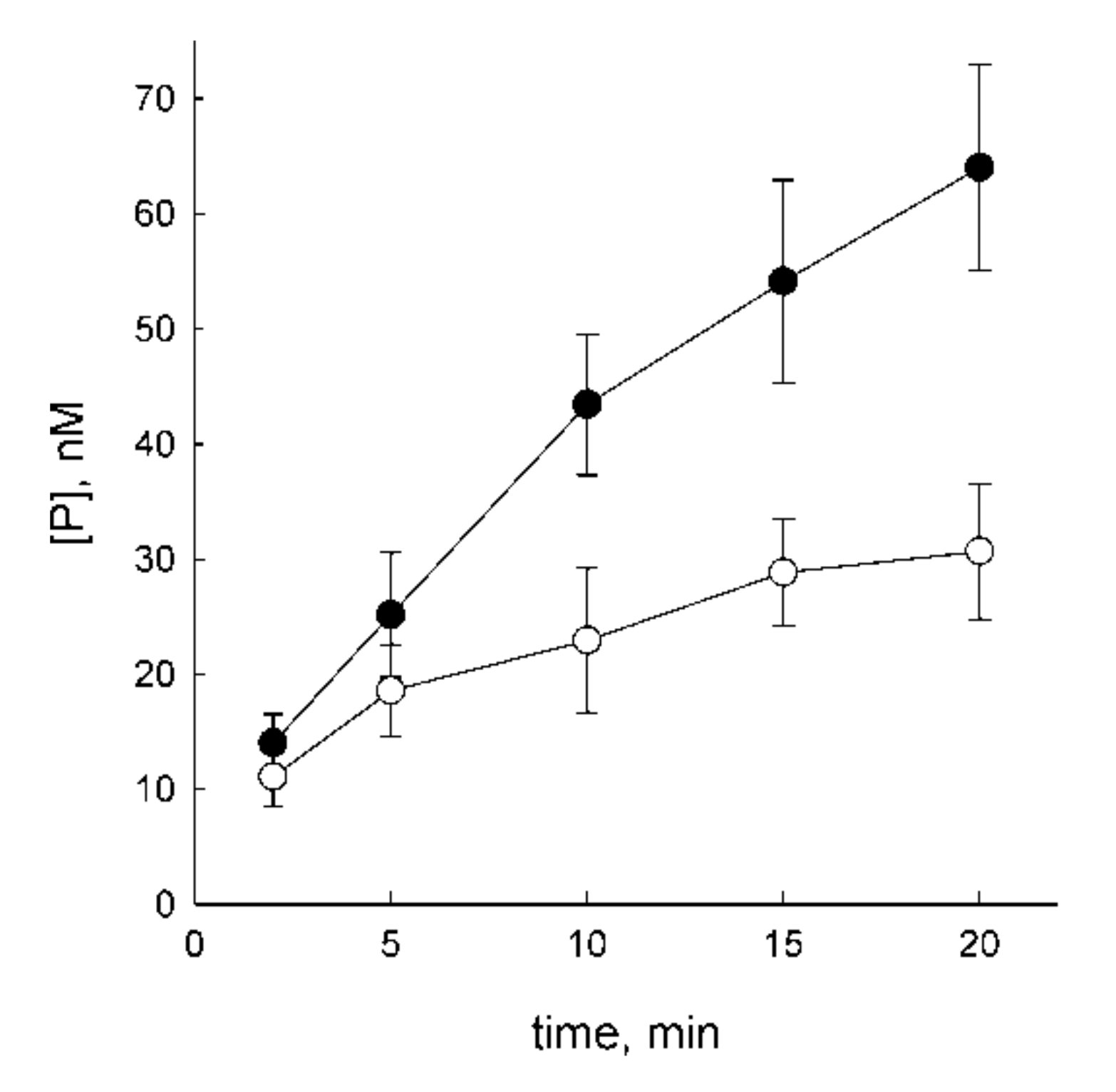
3.1. OGG1 Can Be Displaced by DNA Polymerase β
3.2. OGG1 Can Be Nonspecifically Displaced by the Klenow Fragment
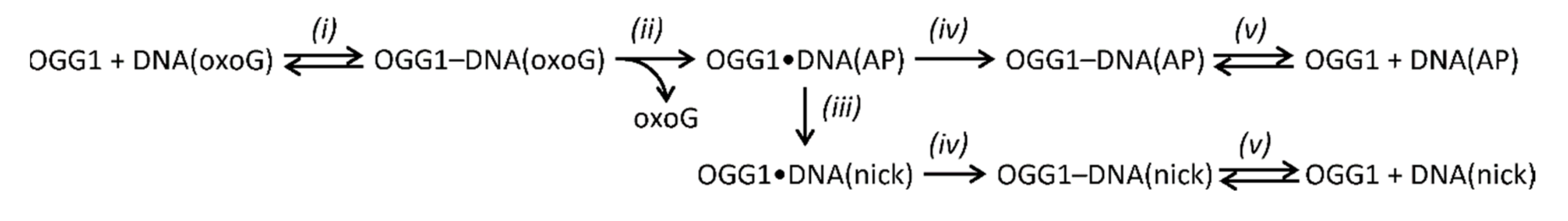
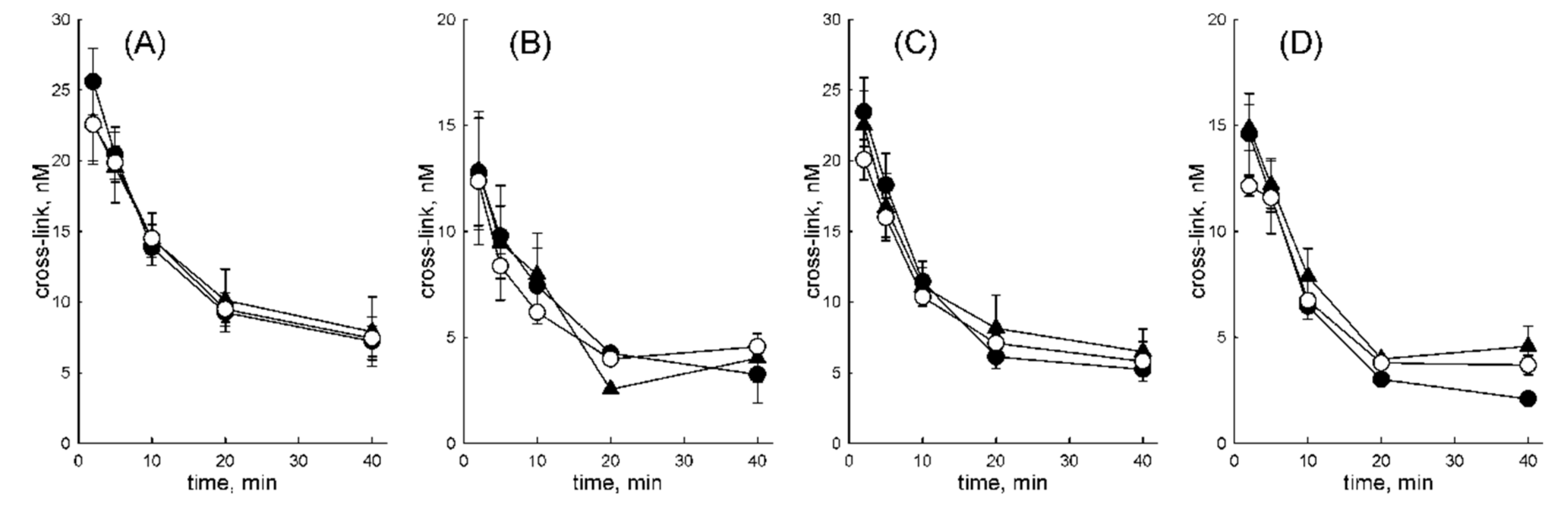
3.3. OGG1 Is Displaced by DNA Polymerases after Hydrolysis of a Schiff Base Intermediate
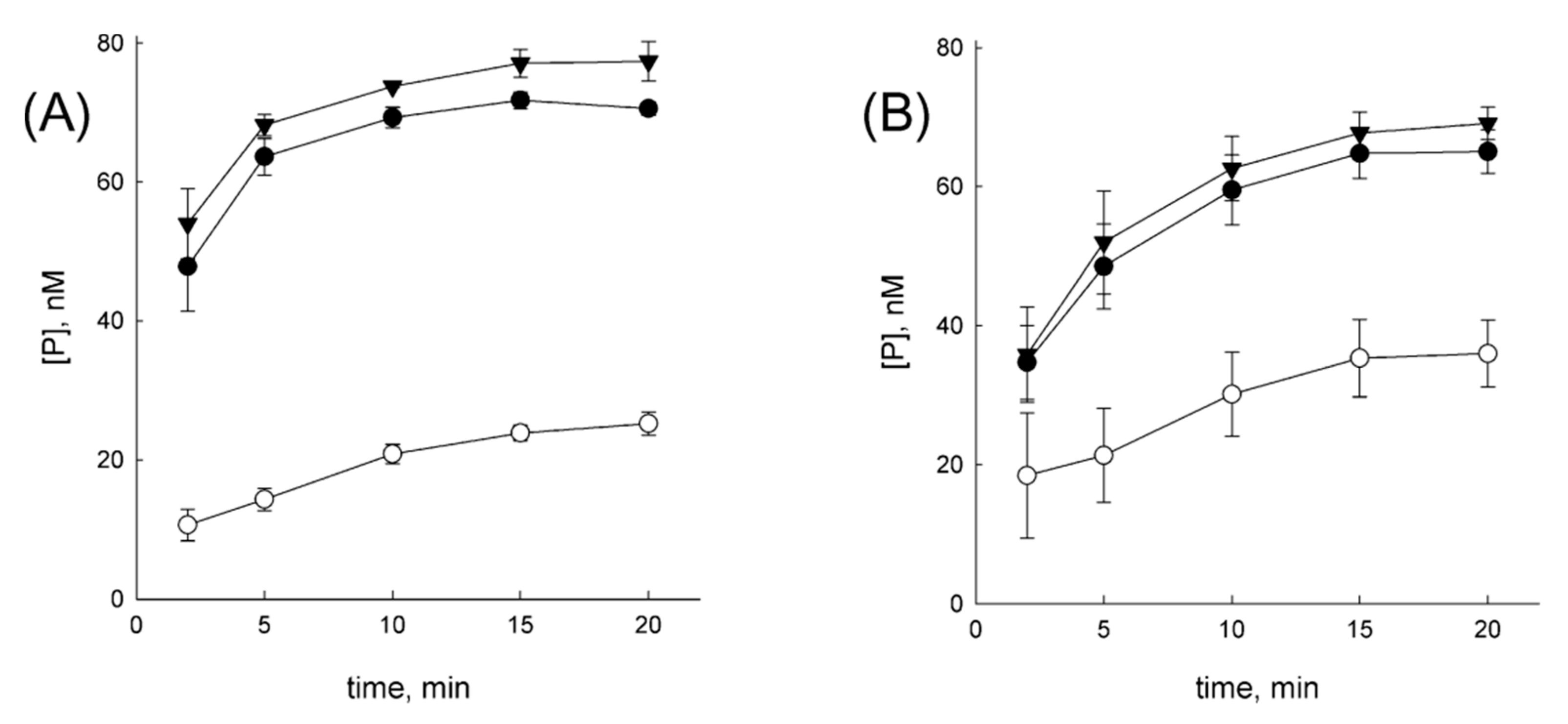
3.4. OGG1 Can Be Displaced by a Nonspecifically Diffusing Protein
3.5. NEIL1 Can Be Displaced by Diffusing Proteins
3.6. Cas9 Is a Barrier for Both Actively Moving and Passively Diffusing Proteins
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bewley, C.A.; Gronenborn, A.M.; Clore, G.M. Minor groove-binding architectural proteins: Structure, function, and DNA recognition. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 105–131. [Google Scholar] [CrossRef]
- Bartholomew, B. Regulating the chromatin landscape: Structural and mechanistic perspectives. Annu. Rev. Biochem. 2014, 83, 671–696. [Google Scholar] [CrossRef] [Green Version]
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, I.J.; Greene, E.C. Molecular traffic jams on DNA. Annu. Rev. Biophys. 2013, 42, 241–263. [Google Scholar] [CrossRef] [Green Version]
- Normanno, D.; Dahan, M.; Darzacq, X. Intra-nuclear mobility and target search mechanisms of transcription factors: A single-molecule perspective on gene expression. Biochim. Biophys. Acta 2012, 1819, 482–493. [Google Scholar] [CrossRef]
- Esadze, A.; Stivers, J.T. Facilitated diffusion mechanisms in DNA base excision repair and transcriptional activation. Chem. Rev. 2018, 118, 11298–11323. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006; p. 1118. [Google Scholar]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Hang, B.; Singer, B. Protein-protein interactions involving DNA glycosylases. Chem. Res. Toxicol. 2003, 16, 1181–1195. [Google Scholar] [CrossRef]
- Endutkin, A.V.; Yudkina, A.V.; Sidorenko, V.S.; Zharkov, D.O. Transient protein–protein complexes in base excision repair. J. Biomol. Struct. Dyn. 2019, 37, 4407–4418. [Google Scholar] [CrossRef]
- Sokhansanj, B.A.; Rodrigue, G.R.; Fitch, J.P.; Wilson, D.M., III. A quantitative model of human DNA base excision repair. I. Mechanistic insights. Nucleic Acids Res. 2002, 30, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair 2007, 6, 317–328. [Google Scholar] [CrossRef]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Shinmura, K.; Yamaguchi, S.; Tani, M.; Seki, S.; Murakami, H.; Nojima, Y.; Yokota, J. Enhancement of OGG1 protein AP lyase activity by increase of APEX protein. Mutat. Res. 2001, 486, 31–40. [Google Scholar] [CrossRef]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef]
- Esadze, A.; Rodriguez, G.; Cravens, S.L.; Stivers, J.T. AP-endonuclease 1 accelerates turnover of human 8-oxoguanine DNA glycosylase by preventing retrograde binding to the abasic-site product. Biochemistry 2017, 56, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Clendenin, W.M.; Wong, D.; Demple, B.; Slupska, M.M.; Chiang, J.-H.; Miller, J.H. Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res. 2001, 29, 743–752. [Google Scholar] [CrossRef]
- Luncsford, P.J.; Manvilla, B.A.; Patterson, D.N.; Malik, S.S.; Jin, J.; Hwang, B.-J.; Gunther, R.; Kalvakolanu, S.; Lipinski, L.J.; Yuan, W.; et al. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair 2013, 12, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.S.; Mol, C.D.; Slupphaug, G.; Bharati, S.; Krokan, H.E.; Tainer, J.A. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998, 17, 5214–5226. [Google Scholar] [CrossRef]
- Waters, T.R.; Gallinari, P.; Jiricny, J.; Swann, P.F. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 1999, 274, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, M.E.; Drohat, A.C. Coordinating the initial steps of base excision repair: Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J. Biol. Chem. 2008, 283, 32680–32690. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, H.; Haushalter, K.A.; Robins, P.; Barnes, D.E.; Verdine, G.L.; Lindahl, T. Excision of deaminated cytosine from the vertebrate genome: Role of the SMUG1 uracil–DNA glycosylase. EMBO J. 2001, 20, 4278–4286. [Google Scholar] [CrossRef] [PubMed]
- Kavli, B.; Sundheim, O.; Akbari, M.; Otterlei, M.; Nilsen, H.; Skorpen, F.; Aas, P.A.; Hagen, L.; Krokan, H.E.; Slupphaug, G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002, 277, 39926–39936. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Zheng, L.; Lee, H.-W.; Bates, S.E.; Federico, L.; Shen, B.; O’Connor, T.R. Human 3-methyladenine-DNA glycosylase: Effect of sequence context on excision, association with PCNA, and stimulation by AP endonuclease. J. Mol. Biol. 2005, 346, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.R.; O’Brien, P.J. Human AP endonuclease 1 stimulates multiple-turnover base excision by alkyladenine DNA glycosylase. Biochemistry 2009, 48, 6022–6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenstein, D.R.; Chan, M.K.; Altamirano, A.; Basu, A.K.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Substrate specificity of human endonuclease III (hNTH1): Effect of human APE1 on hNTH1 activity. J. Biol. Chem. 2003, 278, 9005–9012. [Google Scholar] [CrossRef] [Green Version]
- Miller, H.; Grollman, A.P. Kinetics of DNA polymerase I (Klenow fragment exo–) activity on damaged DNA templates: Effect of proximal and distal template damage on DNA synthesis. Biochemistry 1997, 36, 15336–15342. [Google Scholar] [CrossRef]
- Kumar, A.; Widen, S.G.; Williams, K.R.; Kedar, P.; Karpel, R.L.; Wilson, S.H. Studies of the domain structure of mammalian DNA polymerase β: Identification of a discrete template binding domain. J. Biol. Chem. 1990, 265, 2124–2131. [Google Scholar]
- Freisinger, E.; Grollman, A.P.; Miller, H.; Kisker, C. Lesion (in)tolerance reveals insights into DNA replication fidelity. EMBO J. 2004, 23, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Nevinsky, G.A.; Douglas, K.T.; Fedorova, O.S. Kinetics of substrate recognition and cleavage by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res. 2005, 33, 3919–3931. [Google Scholar] [CrossRef]
- Katafuchi, A.; Nakano, T.; Masaoka, A.; Terato, H.; Iwai, S.; Hanaoka, F.; Ide, H. Differential specificity of human and Escherichia coli endonuclease III and VIII homologues for oxidative base lesions. J. Biol. Chem. 2004, 279, 14464–14471. [Google Scholar] [CrossRef] [Green Version]
- Scaramozzino, N.; Sanz, G.; Crance, J.M.; Saparbaev, M.; Drillien, R.; Laval, J.; Kavli, B.; Garin, D. Characterisation of the substrate specificity of homogeneous vaccinia virus uracil-DNA glycosylase. Nucleic Acids Res. 2003, 31, 4950–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Jinek, M. In vitro enzymology of Cas9. Methods Enzymol. 2014, 546, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.-J.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Bruner, S.D.; Norman, D.P.G.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef]
- Batra, V.K.; Beard, W.A.; Shock, D.D.; Krahn, J.M.; Pedersen, L.C.; Wilson, S.H. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure 2006, 14, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Craggs, T.D.; Sustarsic, M.; Plochowietz, A.; Mosayebi, M.; Kaju, H.; Cuthbert, A.; Hohlbein, J.; Domicevica, L.; Biggin, P.C.; Doye, J.P.K.; et al. Substrate conformational dynamics facilitate structure-specific recognition of gapped DNA by DNA polymerase. Nucleic Acids Res. 2019, 47, 10788–10800. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, J.R.; Mao, C.; Braman, J.C.; Beese, L.S. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature 1998, 391, 304–307. [Google Scholar] [CrossRef]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Specificity of stimulation of human 8-oxoguanine-DNA glycosylase by AP endonuclease. Biochem. Biophys. Res. Commun. 2008, 368, 175–179. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Rosenquist, T.A.; Gerchman, S.E.; Grollman, A.P. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 2000, 275, 28607–28617. [Google Scholar] [CrossRef] [Green Version]
- Yudkina, A.V.; Dvornikova, A.P.; Zharkov, D.O. Variable termination sites of DNA polymerases encountering a DNA–protein cross-link. PLoS ONE 2018, 13, e0198480. [Google Scholar] [CrossRef]
- Beard, W.A.; Osheroff, W.P.; Prasad, R.; Sawaya, M.R.; Jaju, M.; Wood, T.G.; Kraut, J.; Kunkel, T.A.; Wilson, S.H. Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase β. J. Biol. Chem. 1996, 271, 12141–12144. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Kraynov, V.S.; Zhong, X.; Werneburg, B.G.; Tsai, M.-D. DNA polymerase β: Effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem. J. 1998, 331, 79–87. [Google Scholar] [CrossRef]
- Sukhanova, M.; Khodyreva, S.; Lavrik, O. Poly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase β in long patch base excision repair. Mutat. Res. 2010, 685, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.K.; Kedar, P.S.; Stumpo, D.J.; Bertocci, B.; Freedman, J.H.; Samson, L.D.; Wilson, S.H. DNA polymerases β and λ mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS ONE 2010, 5, e12229. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, A.; Gerlt, J.A.; Absalon, M.J.; Stubbe, J.; Cunningham, R.P.; Withka, J.; Bolton, P.H. Stereochemical studies of the β-elimination reactions at aldehydic abasic sites in DNA: Endonuclease III from Escherichia coli, sodium hydroxide, and Lys-Trp-Lys. Biochemistry 1991, 30, 1119–1126. [Google Scholar] [CrossRef]
- Rosenquist, T.A.; Zharkov, D.O.; Grollman, A.P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 7429–7434. [Google Scholar] [CrossRef] [Green Version]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 100. [Google Scholar] [CrossRef] [Green Version]
- Eun, H.-M. DNA Polymerases. In Enzymology Primer for Recombinant DNA Technology; Academic Press: San Diego, CA, USA, 1996; pp. 345–489. [Google Scholar]
- Bjørås, M.; Luna, L.; Johnsen, B.; Hoff, E.; Haug, T.; Rognes, T.; Seeberg, E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997, 16, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Latham, K.A.; Dodson, M.L.; Lloyd, R.S. Studies of the catalytic mechanism of five DNA glycosylases: Probing for enzyme-DNA imino intermediates. J. Biol. Chem. 1995, 270, 19501–19508. [Google Scholar] [CrossRef] [Green Version]
- McCullough, A.K.; Sanchez, A.; Dodson, M.L.; Marapaka, P.; Taylor, J.-S.; Lloyd, R.S. The reaction mechanism of DNA glycosylase/AP lyases at abasic sites. Biochemistry 2001, 40, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.T.; Upton, C.; Higman, M.A.; Niles, E.G.; McFadden, G. A poxvirus-encoded uracil DNA glycosylase is essential for virus viability. J. Virol. 1993, 67, 2503–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanitsa, E.S.; Arps, L.; Traktman, P. Vaccinia virus uracil DNA glycosylase interacts with the A20 protein to form a heterodimeric processivity factor for the viral DNA polymerase. J. Biol. Chem. 2006, 281, 3439–3451. [Google Scholar] [CrossRef]
- Sèle, C.; Gabel, F.; Gutsche, I.; Ivanov, I.; Burmeister, W.P.; Iseni, F.; Tarbouriech, N. Low-resolution structure of vaccinia virus DNA replication machinery. J. Virol. 2013, 87, 1679–1689. [Google Scholar] [CrossRef] [Green Version]
- Schormann, N.; Grigorian, A.; Samal, A.; Krishnan, R.; DeLucas, L.; Chattopadhyay, D. Crystal structure of vaccinia virus uracil-DNA glycosylase reveals dimeric assembly. BMC Struct. Biol. 2007, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef] [Green Version]
- Schomacher, L.; Han, D.; Musheev, M.U.; Arab, K.; Kienhöfer, S.; von Seggern, A.; Niehrs, C. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol. 2016, 23, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Kladova, O.A.; Grin, I.R.; Fedorova, O.S.; Kuznetsov, N.A.; Zharkov, D.O. Conformational dynamics of damage processing by human DNA glycosylase NEIL1. J. Mol. Biol. 2019, 431, 1098–1112. [Google Scholar] [CrossRef]
- Guan, X.; Bai, H.; Shi, G.; Theriot, C.A.; Hazra, T.K.; Mitra, S.; Lu, A.-L. The human checkpoint sensor Rad9–Rad1–Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007, 35, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen: The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Guo, Z.; Gary, R.K.; Hazra, T.K.; Shen, B.; Mitra, S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028–27037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muftuoglu, M.; de Souza-Pinto, N.C.; Dogan, A.; Aamann, M.; Stevnsner, T.; Rybanska, I.; Kirkali, G.; Dizdaroglu, M.; Bohr, V.A. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem. 2009, 284, 9270–9279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theriot, C.A.; Hegde, M.L.; Hazra, T.K.; Mitra, S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair 2010, 9, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Boldogh, I.; Lee, J.W.; Harrigan, J.A.; Hegde, M.L.; Piotrowski, J.; de Souza Pinto, N.; Ramos, W.; Greenberg, M.M.; Hazra, T.K.; et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 2007, 282, 26591–26602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popuri, V.; Croteau, D.L.; Bohr, V.A. Substrate specific stimulation of NEIL1 by WRN but not the other human RecQ helicases. DNA Repair 2010, 9, 636–642. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.M.; Dutta, A.; Sengupta, S.; Mitra, J.; Adhikari, S.; Tomkinson, A.E.; Li, G.-M.; Boldogh, I.; Hazra, T.K.; Mitra, S.; et al. The C-terminal domain (CTD) of human DNA glycosylase NEIL1 is required for forming BERosome repair complex with DNA replication proteins at the replicating genome: Dominant negative function of the CTD. J. Biol. Chem. 2015, 290, 20919–20933. [Google Scholar] [CrossRef] [Green Version]
- Pestryakov, P.; Zharkov, D.O.; Grin, I.; Fomina, E.E.; Kim, E.R.; Hamon, L.; Eliseeva, I.A.; Petruseva, I.O.; Curmi, P.A.; Ovchinnikov, L.P.; et al. Effect of the multifunctional proteins RPA, YB-1, and XPC repair factor on AP site cleavage by DNA glycosylase NEIL1. J. Mol. Recognit. 2012, 25, 224–233. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Moor, N.A.; Naumenko, K.N.; Kutuzov, M.M.; Sukhanova, M.V.; Pestryakov, P.E.; Lavrik, O.I. Y-box-binding protein 1 as a non-canonical factor of base excision repair. Biochim. Biophys. Acta 2016, 1864, 1631–1640. [Google Scholar] [CrossRef]
- Raper, A.T.; Stephenson, A.A.; Suo, Z. Functional insights revealed by the kinetic mechanism of CRISPR/Cas9. J. Am. Chem. Soc. 2018, 140, 2971–2984. [Google Scholar] [CrossRef]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Konigsberg, W.H. RB69 DNA polymerase structure, kinetics, and fidelity. Biochemistry 2014, 53, 2752–2767. [Google Scholar] [CrossRef] [PubMed]
- Huai, C.; Li, G.; Yao, R.; Zhang, Y.; Cao, M.; Kong, L.; Jia, C.; Yuan, H.; Chen, H.; Lu, D.; et al. Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun. 2017, 8, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, J.A.; Cao, F.J.; Lázaro, J.M.; Arias-Gonzalez, J.R.; Valpuesta, J.M.; Carrascosa, J.L.; Salas, M.; Ibarra, B. Mechano-chemical kinetics of DNA replication: Identification of the translocation step of a replicative DNA polymerase. Nucleic Acids Res. 2015, 43, 3643–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, O.; Khamis, H.; Rudnizky, S.; Kaplan, A. The mechano-chemistry of a monomeric reverse transcriptase. Nucleic Acids Res. 2017, 45, 12954–12962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whinn, K.S.; Kaur, G.; Lewis, J.S.; Schauer, G.D.; Mueller, S.H.; Jergic, S.; Maynard, H.; Gan, Z.Y.; Naganbabu, M.; Bruchez, M.P.; et al. Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci. Rep. 2019, 9, 13292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, M.J.; Wilson, S.H. DNA scanning by base excision repair enzymes and implications for pathway coordination. DNA Repair 2018, 71, 101–107. [Google Scholar] [CrossRef]
- Jezewska, M.J.; Galletto, R.; Bujalowski, W. Rat polymerase β binds double-stranded DNA using exclusively the 8-kDa domain. Stoichiometries, intrinsic affinities, and cooperativities. Biochemistry 2003, 42, 5955–5970. [Google Scholar] [CrossRef]
- Howard, M.J.; Rodriguez, Y.; Wilson, S.H. DNA polymerase β uses its lyase domain in a processive search for DNA damage. Nucleic Acids Res. 2017, 45, 3822–3832. [Google Scholar] [CrossRef] [Green Version]
- Lyamichev, V.; Brow, M.A.D.; Dahlberg, J.E. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science 1993, 260, 778–783. [Google Scholar] [CrossRef]
- Xu, Y.; Grindley, N.D.F.; Joyce, C.M. Coordination between the polymerase and 5′-nuclease components of DNA polymerase I of Escherichia coli. J. Biol. Chem. 2000, 275, 20949–20955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezewska, M.J.; Galletto, R.; Bujalowski, W. Dynamics of gapped DNA recognition by human polymerase β. J. Biol. Chem. 2002, 277, 20316–20327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, P.; Bertram, J.G.; O’Donnell, M.; Woodgate, R.; Goodman, M.F. A model for SOS-lesion-targeted mutations in Escherichia coli. Nature 2001, 409, 366–370. [Google Scholar] [CrossRef]
- Goodman, M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002, 71, 17–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dianov, G.L.; O’Neill, P.; Goodhead, D.T. Securing genome stability by orchestrating DNA repair: Removal of radiation-induced clustered lesions in DNA. Bioessays 2001, 23, 745–749. [Google Scholar] [CrossRef]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- David-Cordonnier, M.-H.; Boiteux, S.; O’Neill, P. Efficiency of excision of 8-oxo-guanine within DNA clustered damage by XRS5 nuclear extracts and purified human OGG1 protein. Biochemistry 2001, 40, 11811–11818. [Google Scholar] [CrossRef]
- Éot-Houllier, G.; Gonera, M.; Gasparutto, D.; Giustranti, C.; Sage, E. Interplay between DNA N-glycosylases/AP lyases at multiply damaged sites and biological consequences. Nucleic Acids Res. 2007, 35, 3355–3366. [Google Scholar] [CrossRef] [Green Version]
- Budworth, H.; Matthewman, G.; O’Neill, P.; Dianov, G.L. Repair of tandem base lesions in DNA by human cell extracts generates persisting single-strand breaks. J. Mol. Biol. 2005, 351, 1020–1029. [Google Scholar] [CrossRef]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef] [Green Version]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Szukacsov, V.; Unk, I.; Haracska, L. Human Ape2 protein has a 3′–5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006, 34, 2508–2515. [Google Scholar] [CrossRef]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein–protein interactions. EMBO J. 2001, 20, 6530–6539. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schär, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase β and the XRCC1 protein. EMBO J. 1996, 15, 6662–6670. [Google Scholar] [CrossRef]
- Bhattacharyya, N.; Banerjee, S. A novel role of XRCC1 in the functions of a DNA polymerase β variant. Biochemistry 2001, 40, 9005–9013. [Google Scholar] [CrossRef]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.-H.; Wei, L.; Svilar, A.R.B.D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; Yates, N.A.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat. Commun. 2014, 5, 5513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldecott, K.W.; McKeown, C.K.; Tucker, J.D.; Ljungquist, S.; Thompson, L.H. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol. 1994, 14, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, R.A.; Caldecott, K.W.; Barnes, D.E.; Lindahl, T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry 1997, 36, 5207–5211. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.M.; Wickstead, B.; Cronin, S.; Caldecott, K.W. Role of a BRCT domain in the interaction of DNA ligase III-α with the DNA repair protein XRCC1. Curr. Biol. 1998, 8, 877–880. [Google Scholar] [CrossRef] [Green Version]
- Marsin, S.; Vidal, A.E.; Sossou, M.; Ménissier-de Murcia, J.; Le Page, F.; Boiteux, S.; de Murcia, G.; Radicella, J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003, 278, 44068–44074. [Google Scholar] [CrossRef] [Green Version]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, R.E. The structural basis of XRCC1-mediated DNA repair. DNA Repair 2015, 30, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID 1 | Sequence (5′→3′) | Length | Modification |
|---|---|---|---|
| P | CGAGACCGTCG | 11 | |
| DC | GAGGAAAGAAGCGAAGGAATTCCAGAGC | 28 | |
| DG | GAGGAAAGAAGGGAAGGAATTCCAGAGC | 28 | |
| DU | GAGGAAAGAAGUGAAGGAATTCCAGAGC | 28 | dU |
| DX | GAGGAAAGAAGXGAAGGAATTCCAGAGC | 28 | X = oxoG |
| DCas | GAGGAAACTGATAACTCAATTTGTAAAAAATGGTACTG-AGC | 41 | |
| TC | GCTCTGGAATTCCTTCCCTTCTTTCCTCTCGACGGTCTCG | 40 | |
| TG | GCTCTGGAATTCCTTCGCTTCTTTCCTCTCGACGGTCTCG | 40 | |
| TU | GCTCTGGAATTCCTTCUCTTCTTTCCTCTCGACGGTCTCG | 40 | dU |
| TX | GCTCTGGAATTCCTTCXCTTCTTTCCTCTCGACGGTCTCG | 40 | X = oxoG |
| TCas | GCTCAGTACCATTTTTTACAAATTGAGTTATCAGTTTCC-TCTCGACGGTCTCG | 53 | |
| S2X | CTCTCCCTTCXCTCCTTTCCTCT | 23 | X = oxoG |
| S2Y | CTCTCCCTTCYCTCCTTTCCTCT | 23 | Y = ohU |
| 2S | AGAGGAAAGGAGCGAAGGGAGAG | 23 | |
| S2Cas | CTGATAACTCAATTTGTAAAAAATGGTACTGAGCA | 35 | |
| Cas2S | TGCTCACTACCATTTTTTACAAATTGAGTTATCAG | 35 | |
| M21 | CGAGACCGTCGAGAGGAAACT | 21 | |
| M41 | CGAGACCGTCGAGAGGAAACTGATAACTCAATTTGTAA-AAA | 41 | |
| M47 | CGAGACCGTCGAGAGGAAACTGATAACTCAATTTGTAA-AAAATGGTA | 47 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yudkina, A.V.; Endutkin, A.V.; Diatlova, E.A.; Moor, N.A.; Vokhtantsev, I.P.; Grin, I.R.; Zharkov, D.O. Displacement of Slow-Turnover DNA Glycosylases by Molecular Traffic on DNA. Genes 2020, 11, 866. https://doi.org/10.3390/genes11080866
Yudkina AV, Endutkin AV, Diatlova EA, Moor NA, Vokhtantsev IP, Grin IR, Zharkov DO. Displacement of Slow-Turnover DNA Glycosylases by Molecular Traffic on DNA. Genes. 2020; 11(8):866. https://doi.org/10.3390/genes11080866
Chicago/Turabian StyleYudkina, Anna V., Anton V. Endutkin, Eugenia A. Diatlova, Nina A. Moor, Ivan P. Vokhtantsev, Inga R. Grin, and Dmitry O. Zharkov. 2020. "Displacement of Slow-Turnover DNA Glycosylases by Molecular Traffic on DNA" Genes 11, no. 8: 866. https://doi.org/10.3390/genes11080866
APA StyleYudkina, A. V., Endutkin, A. V., Diatlova, E. A., Moor, N. A., Vokhtantsev, I. P., Grin, I. R., & Zharkov, D. O. (2020). Displacement of Slow-Turnover DNA Glycosylases by Molecular Traffic on DNA. Genes, 11(8), 866. https://doi.org/10.3390/genes11080866