Exploring the lncRNAs Related to Skeletal Muscle Fiber Types and Meat Quality Traits in Pigs

Abstract
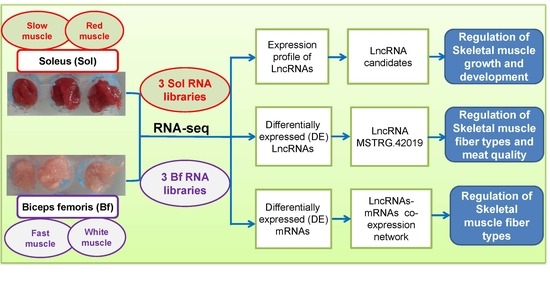
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Samples Collection
2.3. RNA Isolation and LncRNA Library Construction
2.4. Quality Control and Genomic Alignment
2.5. Identification of LncRNAs
2.6. Differentially Expressed Gene Analyses
2.7. Target Prediction of DE LncRNAs and GO and KEGG Enrichment Analyses
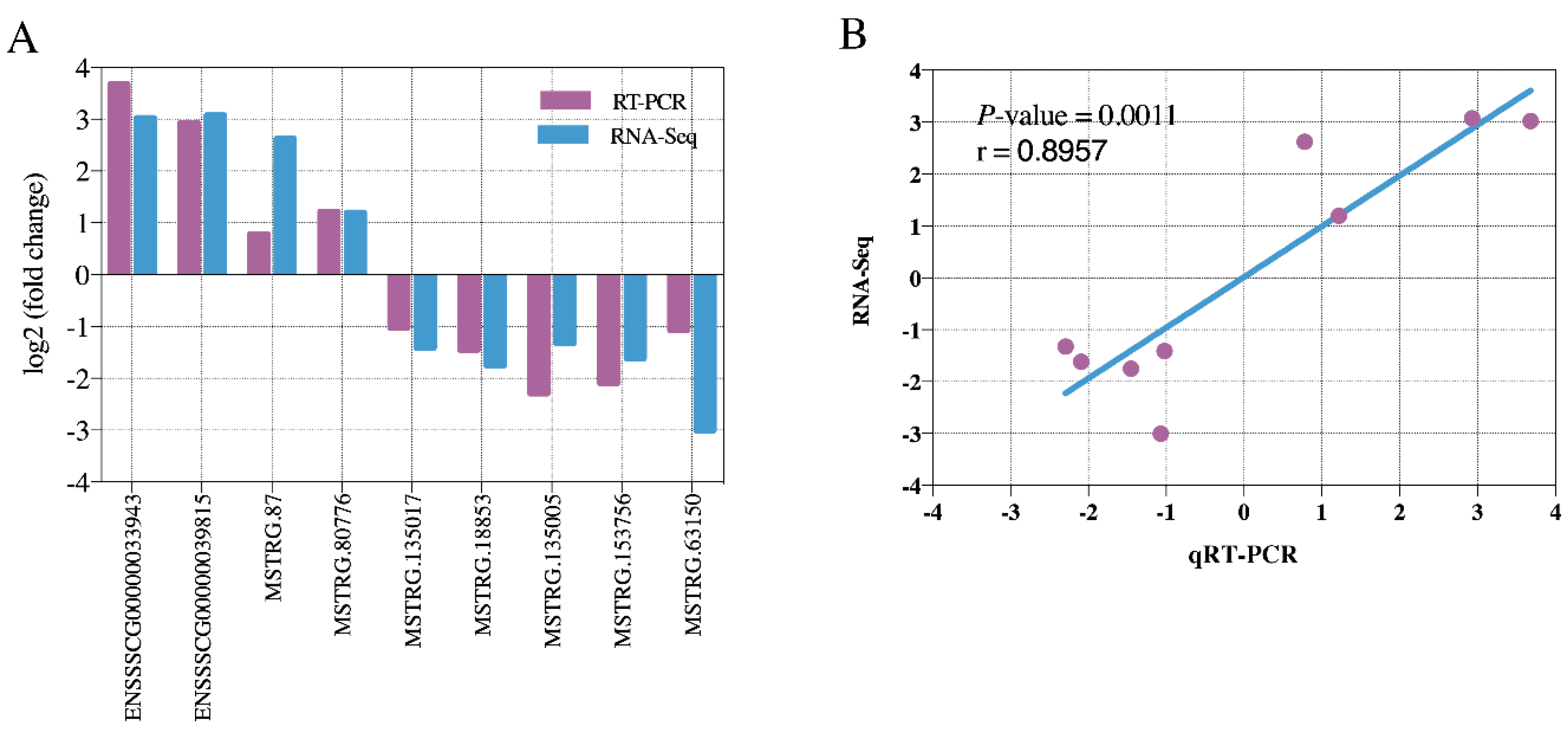
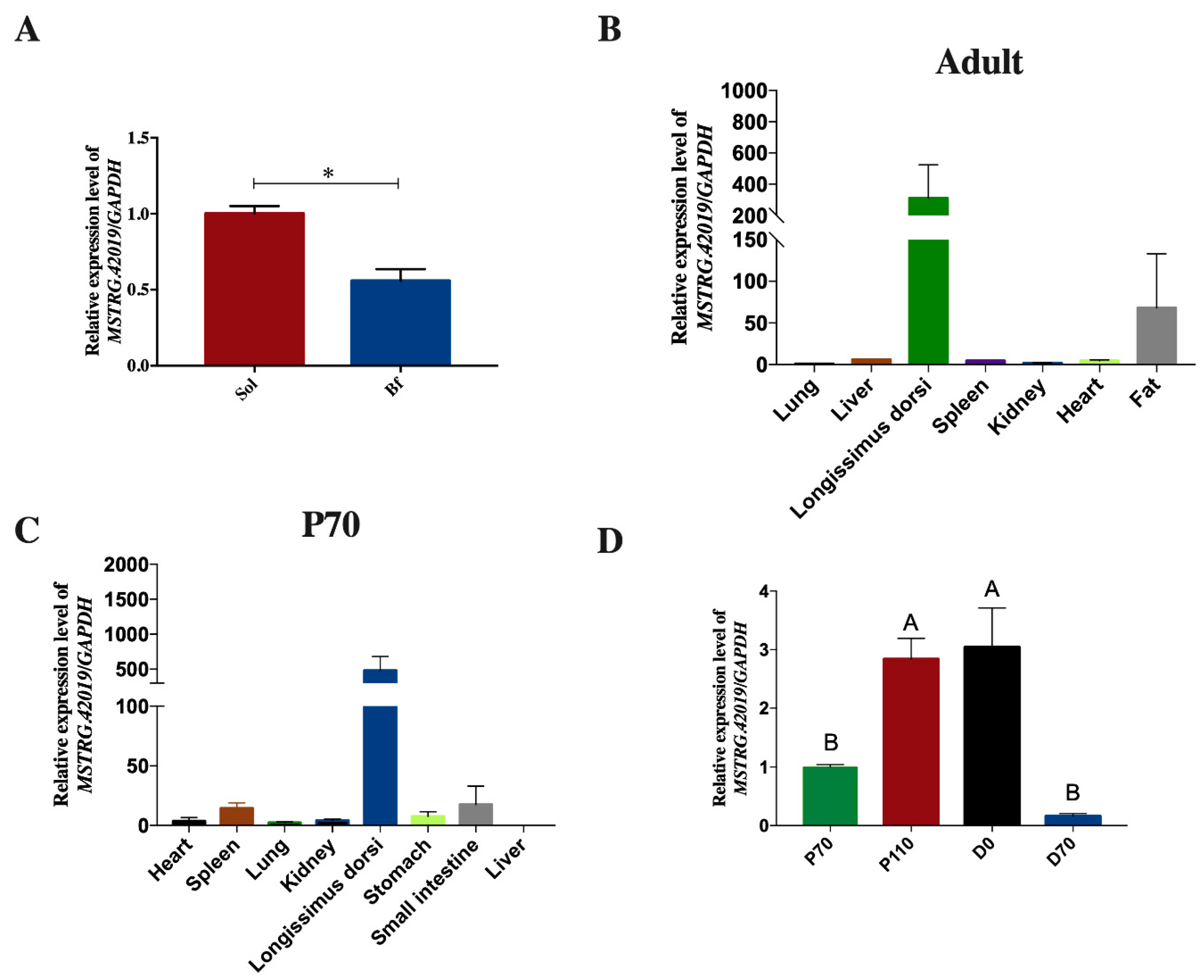
2.8. Expression Patterns of LncRNA MSTRG.42019
2.9. Correlation Analyses of LncRNAs MSTRG.42019 and Meat Quality Traits
2.10. Construction of LncRNA MSTRG.42019–mRNA Network
2.11. Statistical Analyses
3. Results
3.1. Generation of Transcriptome Data
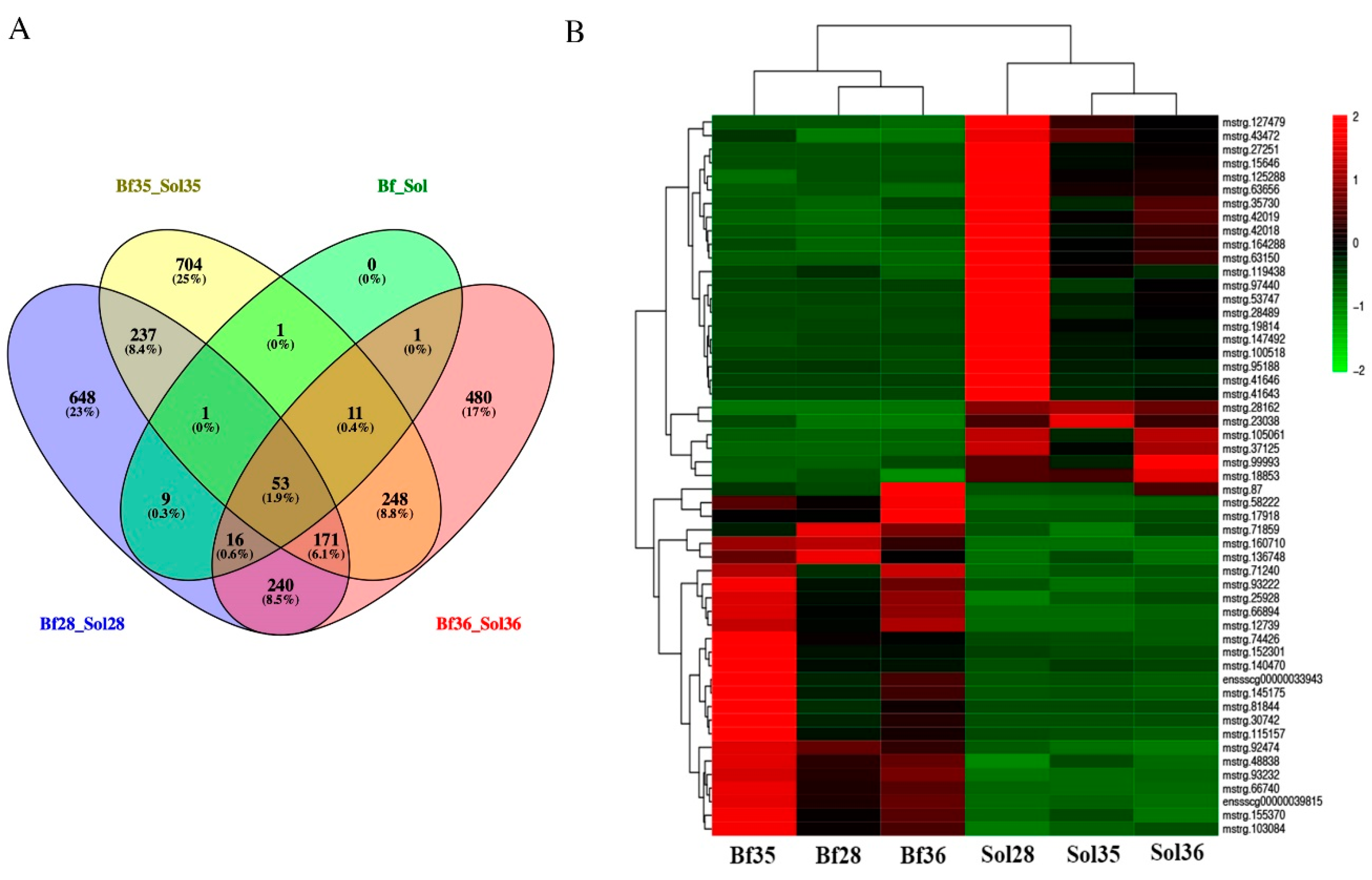
3.2. Novel LncRNAs Prediction and Characteristics Analyses
3.3. Identification of DE LncRNAs and Target Genes Prediction
3.4. GO and KEGG Pathway Enrichment Analyses of Target Genes of DE LncRNAs
3.5. DE LncRNAs Affecting Muscle Fiber Types
3.6. Correlation between LncRNA MSTRG.42019 Expression and Meat Quality Traits
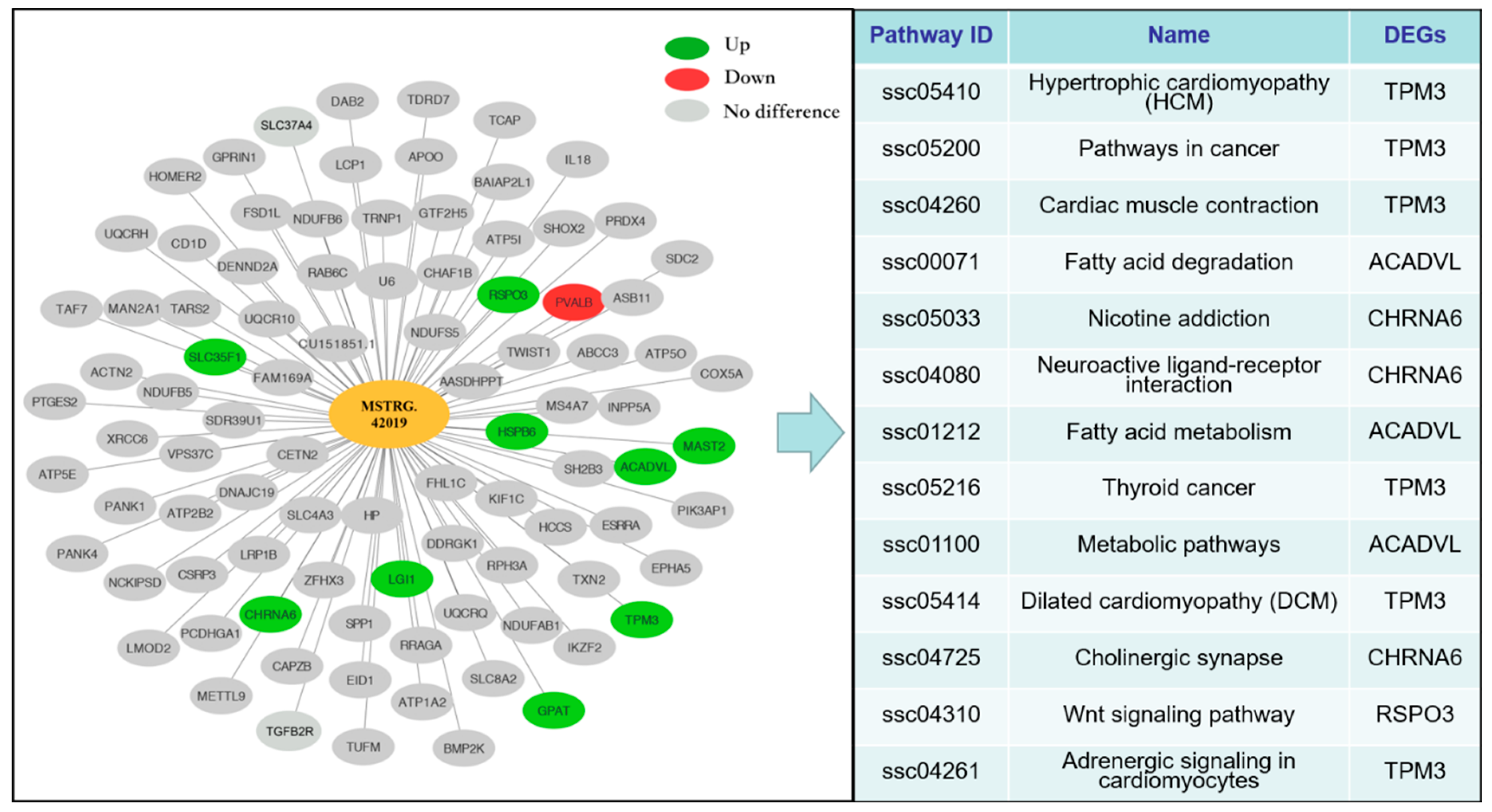
3.7. LncRNA MSTRG.42019–mRNA Interaction Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Gundersen, K. Excitation-transcription coupling in skeletal muscle: The molecular pathways of exercise. Biol. Rev. Camb. Philos. Soc. 2011, 86, 564–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassel-Duby, R.; Olson, E.N. Signaling pathways in skeletal muscle remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, N.L.; Banks, G.B.; Tsang, M.; Margineantu, D.; Gu, H.; Djukovic, D.; Chan, J.; Torres, M.; Liggitt, H.D.; Hirenallur, S.D.; et al. Fnip1 regulates skeletal muscle fiber type specification, fatigue resistance, and susceptibility to muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petchey, L.K.; Risebro, C.A.; Vieira, J.M.; Roberts, T.; Bryson, J.B.; Greensmith, L.; Lythgoe, M.F.; Riley, P.R. Loss of Prox1 in striated muscle causes slow to fast skeletal muscle fiber conversion and dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 9515–9520. [Google Scholar] [CrossRef] [Green Version]
- Boyer, J.G.; Prasad, V.; Song, T.; Lee, D.; Fu, X.; Grimes, K.M.; Sargent, M.A.; Sadayappan, S.; Molkentin, J.D. ERK1/2 signaling induces skeletal muscle slow fiber-type switching and reduces muscular dystrophy disease severity. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.C.; Kim, B.C. The relationship between muscle fiber characteristics, postmortem metabolic rate, and meat quality of pig longissimus dorsi muscle. Meat Sci. 2005, 71, 351–357. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Blais, A.; Tsikitis, M.; Acosta-Alvear, D.; Sharan, R.; Kluger, Y.; Dynlacht, B.D. An initial blueprint for myogenic differentiation. Genes Dev. 2005, 19, 553–569. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.H.; Liu, N.; van Rooij, E.; Olson, E.N. MicroRNA control of muscle development and disease. Curr. Opin. Cell Biol. 2009, 21, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Mok, G.F.; Lozano-Velasco, E.; Munsterberg, A. microRNAs in skeletal muscle development. Semin. Cell Dev. Biol. 2017, 72, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, X.; Sun, H.; Wang, H. Long non-coding RNAs in the regulation of skeletal myogenesis and muscle diseases. Cancer Lett. 2018, 417, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Simionescu-Bankston, A.; Kumar, A. Noncoding RNAs in the regulation of skeletal muscle biology in health and disease. J. Mol. Med. 2016, 94, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, B.K.; Pfeifer, K.; Dutta, A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev. 2014, 28, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, Y.; Bao, X.; Zhu, X.; Kwok, Y.K.; Sun, K.; Chen, X.; Huang, Y.; Jauch, R.; Esteban, M.A.; et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015, 25, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dong, C.; Li, P.; Ren, Z.; Wang, H.; Yu, F.; Ning, C.; Liu, K.; Wei, W.; Huang, R.; et al. Identification of candidate genes associated with porcine meat color traits by genome-wide transcriptome analysis. Sci. Rep. 2016, 6, 35224. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Zhang, X.; Liu, K.; Li, B.; Chao, Z.; Jiang, A.; Li, R.; Li, P.; Liu, H.; Wu, W. Comprehensive Analysis of Porcine Prox1 Gene and Its Relationship with Meat Quality Traits. Animals 2019, 9, 744. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Etwiller, L.; Eddy, S.R.; Griffiths-Jones, S.; Howe, K.L.; Marshall, M.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2002, 30, 276–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Bateman, A.; Finn, R.D. Predicting active site residue annotations in the Pfam database. BMC Bioinform. 2007, 8, 298. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, X.; Zhu, Y.; Zhan, S.; Chao, Z.; Zhong, T.; Guo, J.; Wang, Y.; Li, L.; Zhang, H. Genome-Wide Identification and Characterization of Long Noncoding RNAs of Brown to White Adipose Tissue Transformation in Goats. Cells 2019, 8, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Yotsukura, S.; duVerle, D.; Hancock, T.; Natsume-Kitatani, Y.; Mamitsuka, H. Computational recognition for long non-coding RNA (lncRNA): Software and databases. Brief. Bioinform. 2017, 18, 9–27. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Johnson, S.L.; Gamarra, N.I.; Narlikar, G.J. Mechanisms of ATP-Dependent Chromatin Remodeling Motors. Annu. Rev. Biophys. 2016, 45, 153–181. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Ito, N.; Ruegg, U.T.; Takeda, S. ATP-Induced Increase in Intracellular Calcium Levels and Subsequent Activation of mTOR as Regulators of Skeletal Muscle Hypertrophy. Int. J. Mol. Sci. 2018, 19, 2804. [Google Scholar] [CrossRef] [Green Version]
- Casas, M.; Buvinic, S.; Jaimovich, E. ATP signaling in skeletal muscle: From fiber plasticity to regulation of metabolism. Exerc. Sport Sci. Rev. 2014, 42, 110–116. [Google Scholar] [CrossRef]
- Tucci, S.; Mingirulli, N.; Wehbe, Z.; Dumit, V.I.; Kirschner, J.; Spiekerkoetter, U. Mitochondrial fatty acid biosynthesis and muscle fiber plasticity in very long-chain acyl-CoA dehydrogenase-deficient mice. FEBS Lett. 2018, 592, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Joo, S.T.; Joo, S.H.; Hwang, Y.H. The Relationships between Muscle Fiber Characteristics, Intramuscular Fat Content, and Fatty Acid Compositions in M. longissimus lumborum of Hanwoo Steers. Korean J. Food Sci. Anim. Resour. 2017, 37, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Joo, S.T.; Ryu, Y.C. Skeletal muscle fiber type and myofibrillar proteins in relation to meat quality. Meat Sci. 2010, 86, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Bowker, B.; Zhuang, H. Relationship between water-holding capacity and protein denaturation in broiler breast meat. Poult. Sci. 2015, 94, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Bee, G.; Anderson, A.L.; Lonergan, S.M.; Huff-Lonergan, E. Rate and extent of pH decline affect proteolysis of cytoskeletal proteins and water-holding capacity in pork. Meat Sci. 2007, 76, 359–365. [Google Scholar] [CrossRef]
- Brownridge, P.; de Mello, L.V.; Peters, M.; McLean, L.; Claydon, A.; Cossins, A.R.; Whitfield, P.D.; Young, I.S. Regional variation in parvalbumin isoform expression correlates with muscle performance in common carp (Cyprinus carpio). J. Exp. Biol. 2009, 212, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Pauls, T.L.; Cox, J.A.; Berchtold, M.W. The Ca2+-binding proteins parvalbumin and oncomodulin and their genes: New structural and functional findings. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1996, 1306, 39–54. [Google Scholar] [CrossRef]
- Heizmann, C.W. Parvalbumin, an intracellular calcium-binding protein; distribution, properties and possible roles in mammalian cells. Experientia 1984, 40, 910–921. [Google Scholar] [CrossRef]
- Muntener, M.; Kaser, L.; Weber, J.; Berchtold, M.W. Increase of skeletal muscle relaxation speed by direct injection of parvalbumin cDNA. Proc. Natl. Acad. Sci. USA 1995, 92, 6504–6508. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, B.; Dick, J.; Dhoot, G.; Carroll, S.; Vrbova, G.; Nicotera, P.; Pette, D.; Wyss, A.; Bluethmann, H.; Hunziker, W.; et al. Prolonged contraction-relaxation cycle of fast-twitch muscles in parvalbumin knockout mice. Am. J. Physiol. 1999, 276, C395–C403. [Google Scholar] [CrossRef]
- Takeuchi, K.; Reue, K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1195–E1209. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, A.; Lambert, G.; Ernest, S.; Dutrieux, F.X.; Coulpier, F.; Lemoine, S.; Lobbardi, R.; Rosa, F.M. Quaking RNA-Binding Proteins Control Early Myofibril Formation by Modulating Tropomyosin. Dev. Cell 2017, 42, 527–541.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Couturier, A.; Kubens, J.F.; Most, E.; Mooren, F.C.; Kruger, K.; Ringseis, R.; Eder, K. Niacin supplementation induces type II to type I muscle fiber transition in skeletal muscle of sheep. Acta Vet. Scand. 2013, 55, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Li, B.; Jiang, A.; Cao, Y.; Hou, L.; Zhang, Z.; Zhang, X.; Liu, H.; Kim, K.-H.; Wu, W. Exploring the lncRNAs Related to Skeletal Muscle Fiber Types and Meat Quality Traits in Pigs. Genes 2020, 11, 883. https://doi.org/10.3390/genes11080883
Li R, Li B, Jiang A, Cao Y, Hou L, Zhang Z, Zhang X, Liu H, Kim K-H, Wu W. Exploring the lncRNAs Related to Skeletal Muscle Fiber Types and Meat Quality Traits in Pigs. Genes. 2020; 11(8):883. https://doi.org/10.3390/genes11080883
Chicago/Turabian StyleLi, Rongyang, Bojiang Li, Aiwen Jiang, Yan Cao, Liming Hou, Zengkai Zhang, Xiying Zhang, Honglin Liu, Kee-Hong Kim, and Wangjun Wu. 2020. "Exploring the lncRNAs Related to Skeletal Muscle Fiber Types and Meat Quality Traits in Pigs" Genes 11, no. 8: 883. https://doi.org/10.3390/genes11080883