Pathological Consequences of Hepatic mTORC1 Dysregulation



{kind=link}
{kind=link}
Abstract
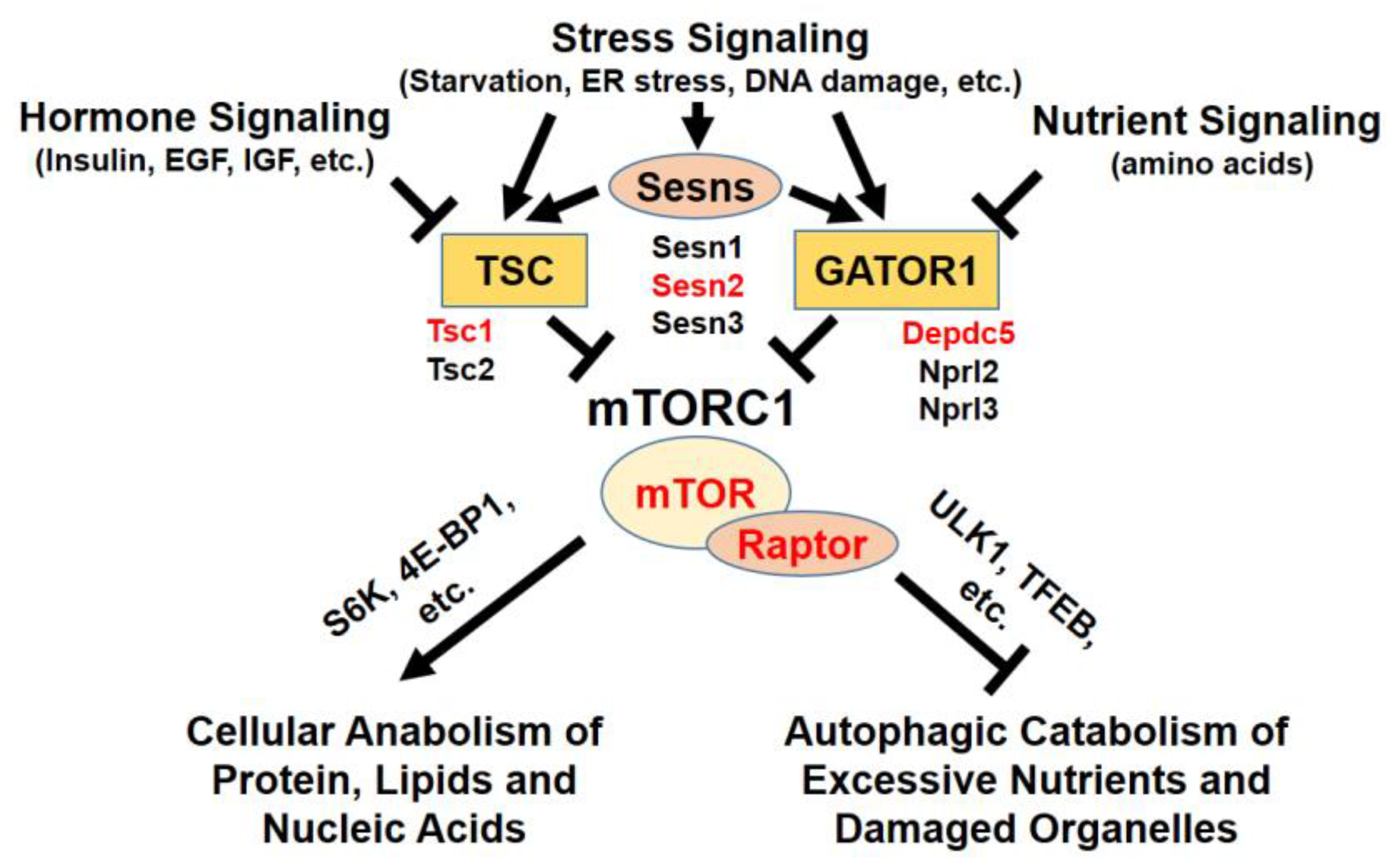
1. Introduction: mTOR Complexes
2. Regulation of mTORC1 by Environmental Inputs
2.1. Insulin/Growth Factor-Dependent Control
2.2. Energy-Dependent Control
2.3. Amino Acid-Dependent Control
2.4. Environmental Stress-Dependent Control
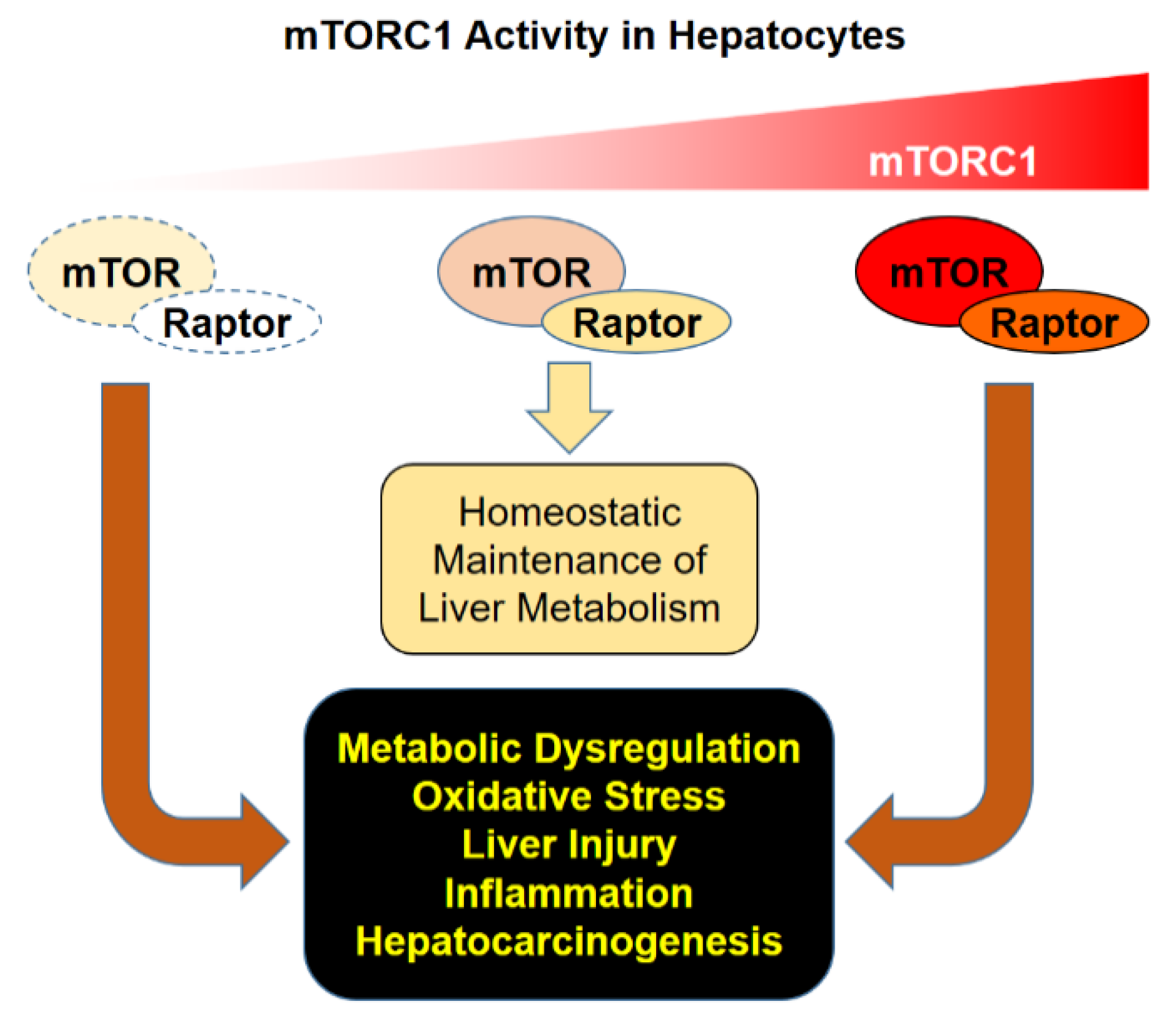
3. Pathological Consequences of mTORC1 Dysregulation in Liver
3.1. Ablation of mTOR
3.2. Ablation of mTORC1
3.3. Concurrent Ablation of Both mTORC1 and Autophagy
3.4. Upregulation of Insulin/Growth Factor Signaling on mTORC1
3.5. Upregulation of Amino Acid Signaling on mTORC1
3.6. Hyperactivation of mTORC1 through Both Growth Factor and Nutrient Pathways
3.7. Ablation of Stress-Dependent mTORC1 Regulation Mechanisms
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Prouteau, M.; Kusmider, B.; Loewith, R. TORC2 Structure and Function. Trends. Biochem. Sci. 2016, 41, 532–545. [Google Scholar] [CrossRef]
- Smithson, L.J.; Gutmann, D.H. Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev. 2016, 30, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Ray, C.; Fox, A.L.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci. Adv. 2018, 4, eaao5838. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Haidar, F.S.; Fox, A.L.; Ray, C.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. mEAK-7 Forms an Alternative mTOR Complex with DNA-PKcs in Human Cancer. iScience 2019, 17, 190–207. [Google Scholar] [CrossRef]
- Harwood, F.C.; Klein Geltink, R.I.; O’Hara, B.P.; Cardone, M.; Janke, L.; Finkelstein, D.; Entin, I.; Paul, L.; Houghton, P.J.; Grosveld, G.C. ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci. Adv. 2018, 4, eaar3938. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends. Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Gonzalez, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef]
- Wiza, C.; Nascimento, E.B.; Ouwens, D.M. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1453–E1460. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Hardie, D.G.; Lin, S.C. AMP-activated protein kinase-not just an energy sensor. F1000Research 2017, 6, 1724. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Kim, M.; Lee, J.H. Identification of an AMPK phosphorylation site in Drosophila TSC2 (gigas) that regulate cell growth. Int. J. Mol. Sci. 2015, 16, 7015–7026. [Google Scholar] [CrossRef]
- Dokudovskaya, S.; Rout, M.P. SEA you later alli-GATOR--a dynamic regulator of the TORC1 stress response pathway. J. Cell Sci. 2015, 128, 2219–2228. [Google Scholar] [CrossRef]
- Panchaud, N.; Peli-Gulli, M.P.; De Virgilio, C. SEACing the GAP that nEGOCiates TORC1 activation: Evolutionary conservation of Rag GTPase regulation. Cell Cycle 2013, 12, 2948–2952. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef]
- Meng, D.; Yang, Q.; Wang, H.; Melick, C.H.; Navlani, R.; Frank, A.R.; Jewell, J.L. Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J. Biol. Chem. 2020, 295, 2890–2899. [Google Scholar] [CrossRef]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab. 2019, 29, 192–201.e7. [Google Scholar] [CrossRef]
- Levine, A.J.; Feng, Z.; Mak, T.W.; You, H.; Jin, S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006, 20, 267–275. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef]
- Kim, J.S.; Ro, S.H.; Kim, M.; Park, H.W.; Semple, I.A.; Park, H.; Cho, U.S.; Wang, W.; Guan, K.L.; Karin, M.; et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef]
- Kim, H.; An, S.; Ro, S.H.; Teixeira, F.; Park, G.J.; Kim, C.; Cho, C.S.; Kim, J.S.; Jakob, U.; Lee, J.H.; et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains. Nat. Commun. 2015, 6, 10025. [Google Scholar] [CrossRef]
- Bruning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef]
- Park, H.W.; Park, H.; Ro, S.H.; Jang, I.; Semple, I.A.; Kim, D.N.; Kim, M.; Nam, M.; Zhang, D.; Yin, L.; et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat. Commun. 2014, 5, 4233. [Google Scholar] [CrossRef]
- Saveljeva, S.; Cleary, P.; Mnich, K.; Ayo, A.; Pakos-Zebrucka, K.; Patterson, J.B.; Logue, S.E.; Samali, A. Endoplasmic reticulum stress-mediated induction of SESTRIN 2 potentiates cell survival. Oncotarget 2016, 7, 12254–12266. [Google Scholar] [CrossRef]
- Jegal, K.H.; Park, S.M.; Cho, S.S.; Byun, S.H.; Ku, S.K.; Kim, S.C.; Ki, S.H.; Cho, I.J. Activating transcription factor 6-dependent sestrin 2 induction ameliorates ER stress-mediated liver injury. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1295–1307. [Google Scholar] [CrossRef]
- Misiewicz, M.; Dery, M.A.; Foveau, B.; Jodoin, J.; Ruths, D.; LeBlanc, A.C. Identification of a novel endoplasmic reticulum stress response element regulated by XBP1. J. Biol. Chem. 2013, 288, 20378–20391. [Google Scholar] [CrossRef]
- Heberle, A.M.; Prentzell, M.T.; van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular mechanisms of mTOR regulation by stress. Mol. Cell Oncol. 2015, 2, e970489. [Google Scholar] [CrossRef]
- Weisend, C.M.; Kundert, J.A.; Suvorova, E.S.; Prigge, J.R.; Schmidt, E.E. Cre activity in fetal albCre mouse hepatocytes: Utility for developmental studies. Genesis 2009, 47, 789–792. [Google Scholar]
- Li, Z.; Zhang, J.; Mulholland, M.; Zhang, W. mTOR activation protects liver from ischemia/reperfusion-induced injury through NF-kappaB pathway. FASEB J. 2017, 31, 3018–3026. [Google Scholar] [CrossRef]
- Kim, D.H.; Sabatini, D.M. Raptor and mTOR: Subunits of a nutrient-sensitive complex. Curr. Top. Microbiol. Immunol. 2004, 279, 259–270. [Google Scholar]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mTOR complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR Complex 1 Regulates Lipin 1 Localization to Control the SREBP Pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Hu, Y.; Semova, I.; Sun, X.; Kang, H.; Chahar, S.; Hollenberg, A.N.; Masson, D.; Hirschey, M.D.; Miao, J.; Biddinger, S.B. Fructose and glucose can regulate mammalian target of rapamycin complex 1 and lipogenic gene expression via distinct pathways. J. Biol. Chem. 2018, 293, 2006–2014. [Google Scholar] [CrossRef]
- Quinn, W.J., 3rd; Wan, M.; Shewale, S.V.; Gelfer, R.; Rader, D.J.; Birnbaum, M.J.; Titchenell, P.M. mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J. Clin. Investig. 2017, 127, 4207–4215. [Google Scholar] [CrossRef]
- Kim, K.; Qiang, L.; Hayden, M.S.; Sparling, D.P.; Purcell, N.H.; Pajvani, U.B. mTORC1-independent Raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat. Commun. 2016, 7, 10255. [Google Scholar] [CrossRef]
- Umemura, A.; Park, E.J.; Taniguchi, K.; Lee, J.H.; Shalapour, S.; Valasek, M.A.; Aghajan, M.; Nakagawa, H.; Seki, E.; Hall, M.N.; et al. Liver Damage, Inflammation, and Enhanced Tumorigenesis after Persistent mTORC1 Inhibition. Cell Metab. 2014, 20, 133–144. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Ni, H.M.; Chao, X.; Yang, H.; Deng, F.; Wang, S.; Bai, Q.; Qian, H.; Cui, Y.; Cui, W.; Shi, Y.; et al. Dual Roles of Mammalian Target of Rapamycin in Regulating Liver Injury and Tumorigenesis in Autophagy-Defective Mouse Liver. Hepatology 2019, 70, 2142–2155. [Google Scholar] [CrossRef]
- Papa, A.; Pandolfi, P.P. The PTEN-PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef]
- Watanabe, S.; Horie, Y.; Kataoka, E.; Sato, W.; Dohmen, T.; Ohshima, S.; Goto, T.; Suzuki, A. Non-alcoholic steatohepatitis and hepatocellular carcinoma: Lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. 1), S96–S100. [Google Scholar] [CrossRef]
- Watanabe, S.; Horie, Y.; Suzuki, A. Hepatocyte-specific Pten-deficient mice as a novel model for nonalcoholic steatohepatitis and hepatocellular carcinoma. Hepatol. Res. 2005, 33, 161–166. [Google Scholar] [CrossRef]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.H.; et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef]
- Menon, S.; Yecies, J.L.; Zhang, H.H.; Howell, J.J.; Nicholatos, J.; Harputlugil, E.; Bronson, R.T.; Kwiatkowski, D.J.; Manning, B.D. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci. Signal. 2012, 5, ra24. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Malla, R.; Wang, Y.; Chan, W.K.; Tiwari, A.K.; Faridi, J.S. Genetic ablation of PRAS40 improves glucose homeostasis via linking the AKT and mTOR pathways. Biochem. Pharmacol. 2015, 96, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Kenerson, H.L.; Yeh, M.M.; Kazami, M.; Jiang, X.; Riehle, K.J.; McIntyre, R.L.; Park, J.O.; Kwon, S.; Campbell, J.S.; Yeung, R.S. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology 2013, 144, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef]
- Cho, C.S.; Kowalsky, A.H.; Namkoong, S.; Park, S.R.; Wu, S.; Kim, B.; James, A.; Gu, B.; Semple, I.A.; Tohamy, M.A.; et al. Concurrent activation of growth factor and nutrient arms of mTORC1 induces oxidative liver injury. Cell Discov. 2019, 5, 60. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef]
- Ho, A.; Cho, C.S.; Namkoong, S.; Cho, U.S.; Lee, J.H. Biochemical Basis of Sestrin Physiological Activities. Trends. Biochem. Sci. 2016, 41, 621–632. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Talukdar, S.; Park, E.J.; Park, H.; Park, H.-W.; Bandyopadhyay, G.; Li, N.; Aghajan, M.; Jang, I.; et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012, 16, 311–321. [Google Scholar] [CrossRef]
- Tao, R.; Xiong, X.; Liangpunsakul, S.; Dong, X.C. Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes 2015, 64, 1211–1223. [Google Scholar] [CrossRef]
- Kowalsky, A.H.; Namkoong, S.; Mettetal, E.; Park, H.W.; Kazyken, D.; Fingar, D.C.; Lee, J.H. The GATOR2-mTORC2 axis mediates Sestrin2-induced AKT Ser/Thr kinase activation. J. Biol. Chem. 2020, 295, 1769–1780. [Google Scholar] [CrossRef]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Kim, H.G.; Zhong, X.; Dong, C.; Zhang, B.; Fang, Z.; Zhang, Y.; Lu, X.; Saxena, R.; Liu, Y.; et al. Sestrin 3 Protects Against Diet-Induced Nonalcoholic Steatohepatitis in Mice Through Suppression of Transforming Growth Factor β Signal Transduction. Hepatology 2020, 71, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Ferrin, G.; Guerrero, M.; Amado, V.; Rodriguez-Peralvarez, M.; De la Mata, M. Activation of mTOR Signaling Pathway in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1266. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, C.-S.; Kowalsky, A.H.; Lee, J.H. Pathological Consequences of Hepatic mTORC1 Dysregulation. Genes 2020, 11, 896. https://doi.org/10.3390/genes11080896
Cho C-S, Kowalsky AH, Lee JH. Pathological Consequences of Hepatic mTORC1 Dysregulation. Genes. 2020; 11(8):896. https://doi.org/10.3390/genes11080896
Chicago/Turabian StyleCho, Chun-Seok, Allison Ho Kowalsky, and Jun Hee Lee. 2020. "Pathological Consequences of Hepatic mTORC1 Dysregulation" Genes 11, no. 8: 896. https://doi.org/10.3390/genes11080896
APA StyleCho, C.-S., Kowalsky, A. H., & Lee, J. H. (2020). Pathological Consequences of Hepatic mTORC1 Dysregulation. Genes, 11(8), 896. https://doi.org/10.3390/genes11080896