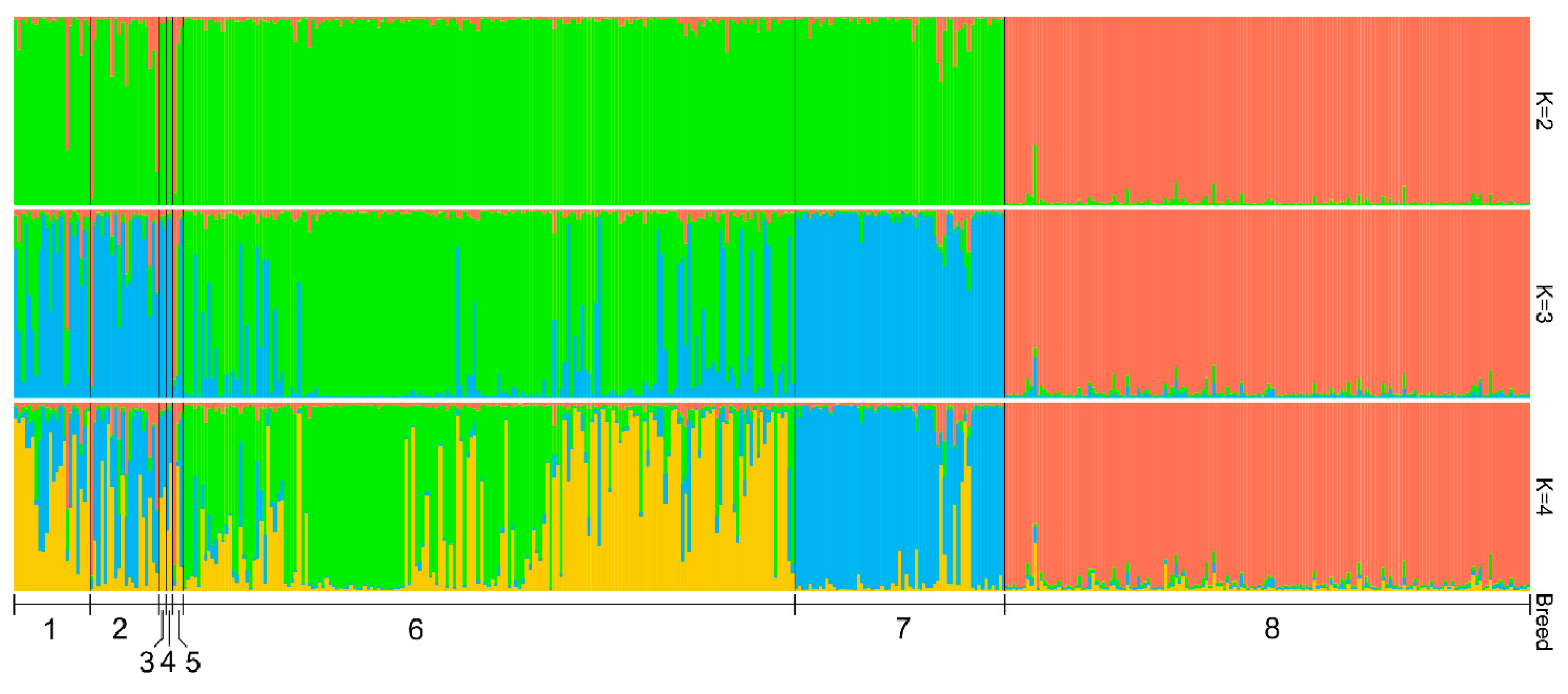
Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis

 ,
, 
 ,
, 
Abstract
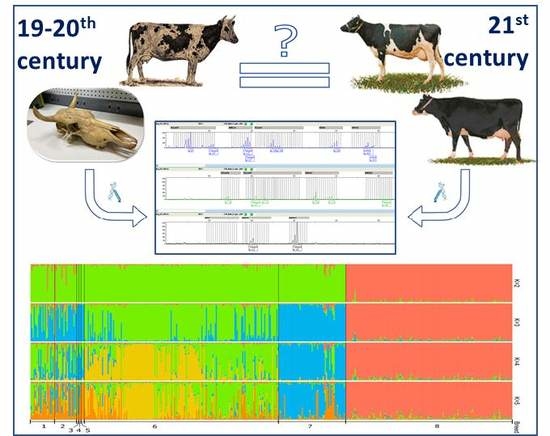
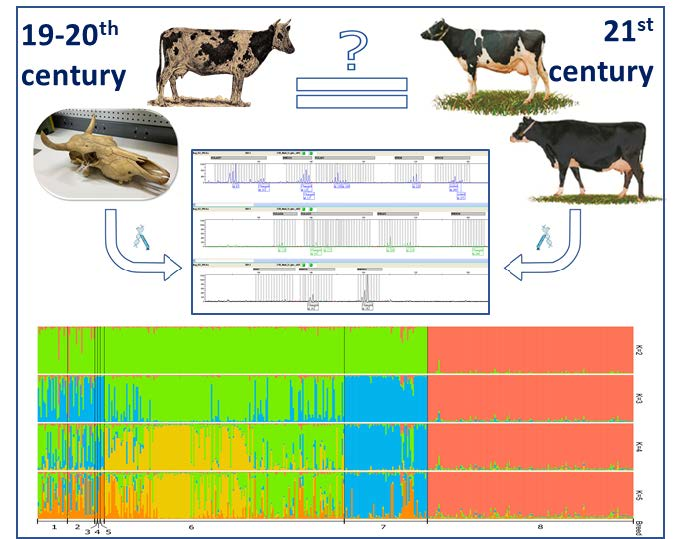{kind=link}
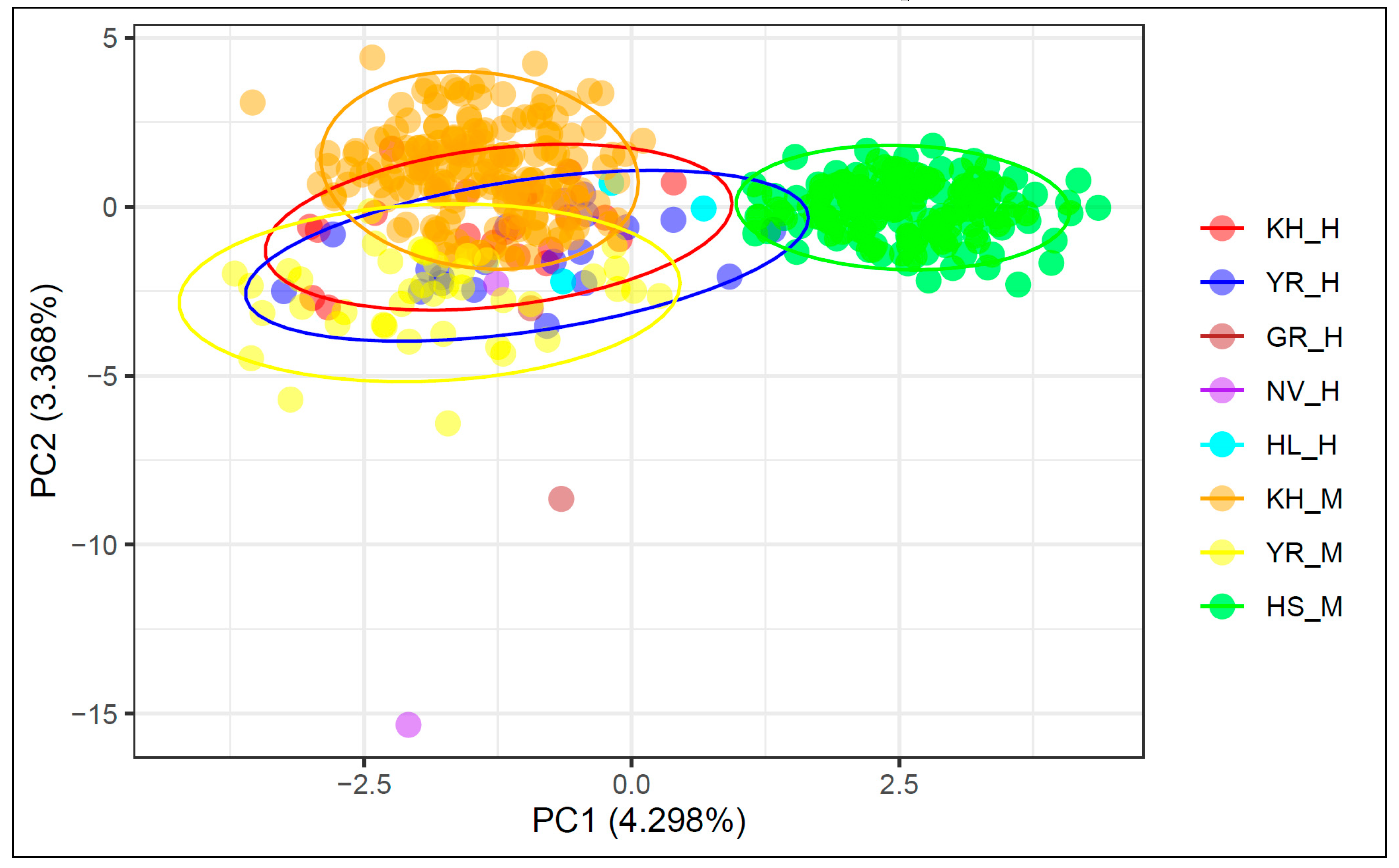{kind=link}
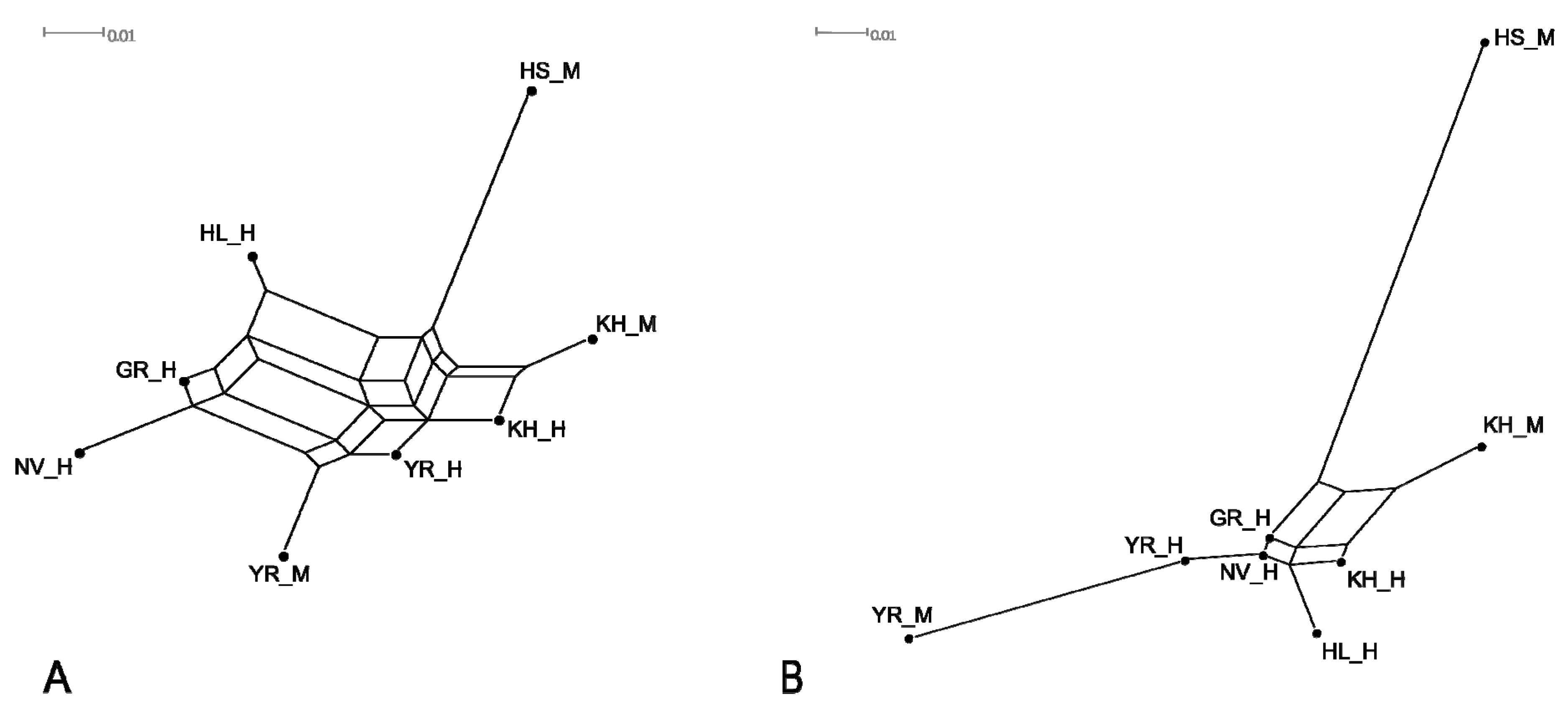{kind=link}
{kind=link}
Share and Cite
Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V.; Mishina, A.I.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.I.; Petrikeeva, L.V.; Chinarov, R.Y.; Brem, G.; et al. Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis. Genes 2020, 11, 940. https://doi.org/10.3390/genes11080940
Abdelmanova AS, Kharzinova VR, Volkova VV, Mishina AI, Dotsev AV, Sermyagin AA, Boronetskaya OI, Petrikeeva LV, Chinarov RY, Brem G, et al. Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis. Genes. 2020; 11(8):940. https://doi.org/10.3390/genes11080940
Chicago/Turabian StyleAbdelmanova, Alexandra S., Veronika R. Kharzinova, Valeria V. Volkova, Arina I. Mishina, Arsen V. Dotsev, Alexander A. Sermyagin, Oxana I. Boronetskaya, Lidia V. Petrikeeva, Roman Yu Chinarov, Gottfried Brem, and et al. 2020. "Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis" Genes 11, no. 8: 940. https://doi.org/10.3390/genes11080940
APA StyleAbdelmanova, A. S., Kharzinova, V. R., Volkova, V. V., Mishina, A. I., Dotsev, A. V., Sermyagin, A. A., Boronetskaya, O. I., Petrikeeva, L. V., Chinarov, R. Y., Brem, G., & Zinovieva, N. A. (2020). Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis. Genes, 11(8), 940. https://doi.org/10.3390/genes11080940