Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic

 , , , , ,
, , , , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Real-Time Polymerase Chain Reaction
2.3. Tiling-Based Polymerase Chain Reaction
2.4. Library Preparation and Next-Generation Sequencing
2.4.1. TruSeq Nano DNA Library Preparation
2.4.2. NEBNext Ultra II DNA Library Preparation
2.4.3. Nextera DNA Flex Library Preparation
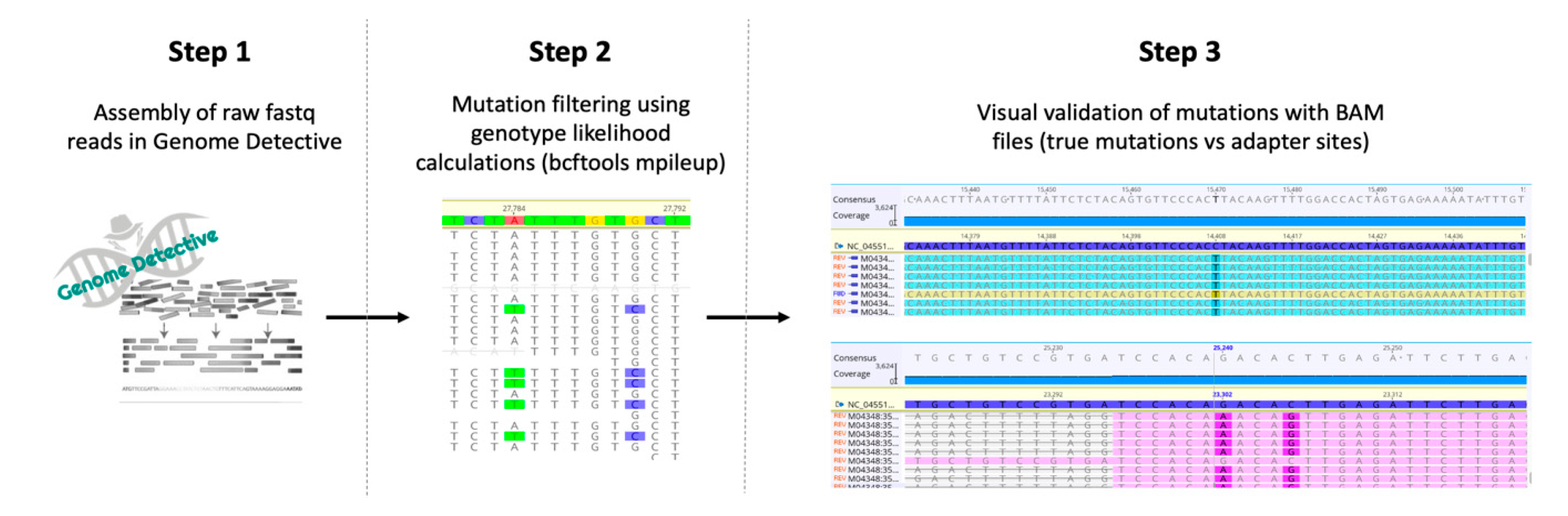
2.5. Data Analysis
3. Results
3.1. Sample Characteristics
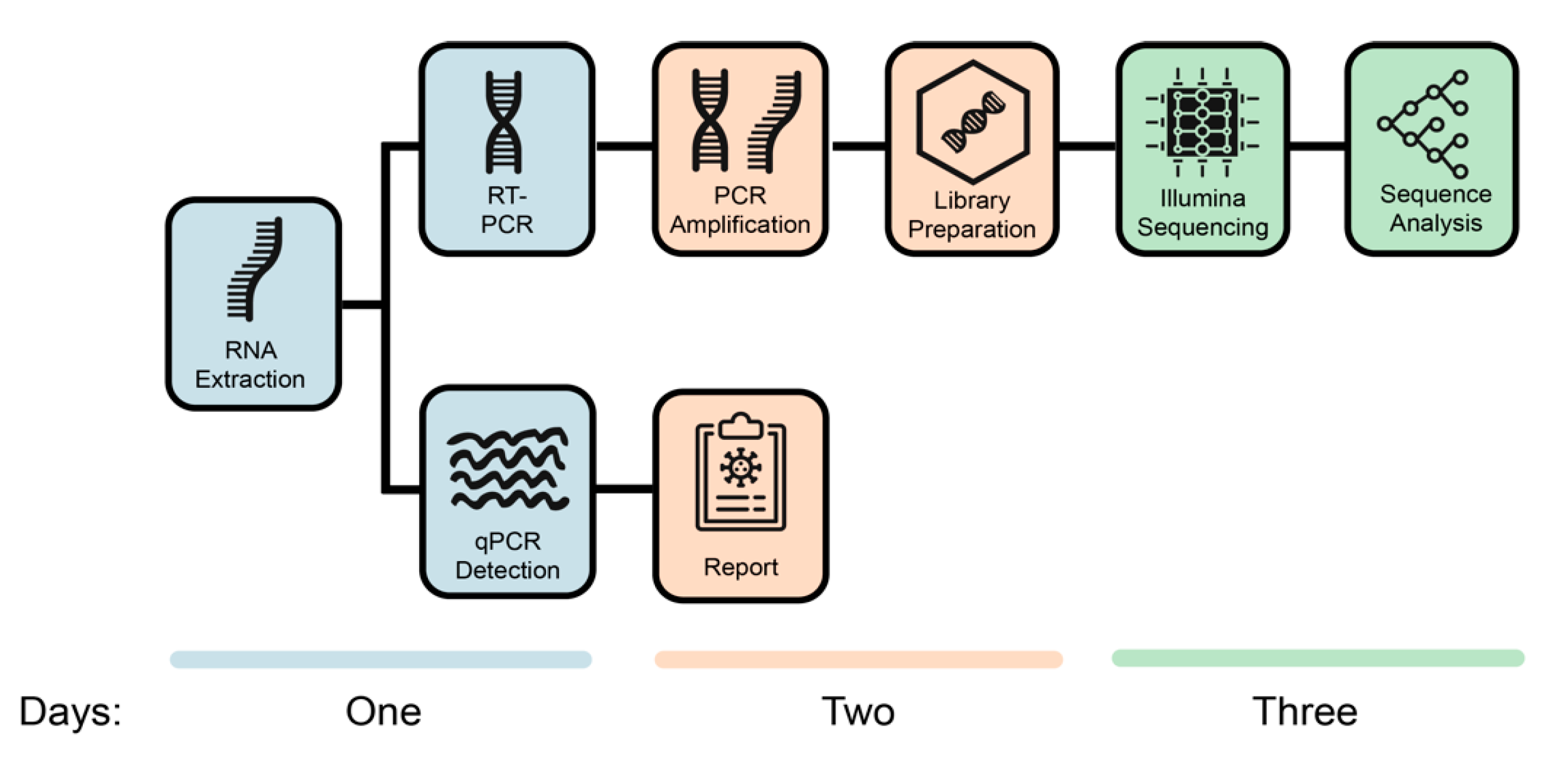
3.2. SARS-CoV-2 Sequencing with Limited Reagents and Unavailability of Stock during a Pandemic in a Country in Lockdown
3.3. SARS-CoV-2 Genome Assembly
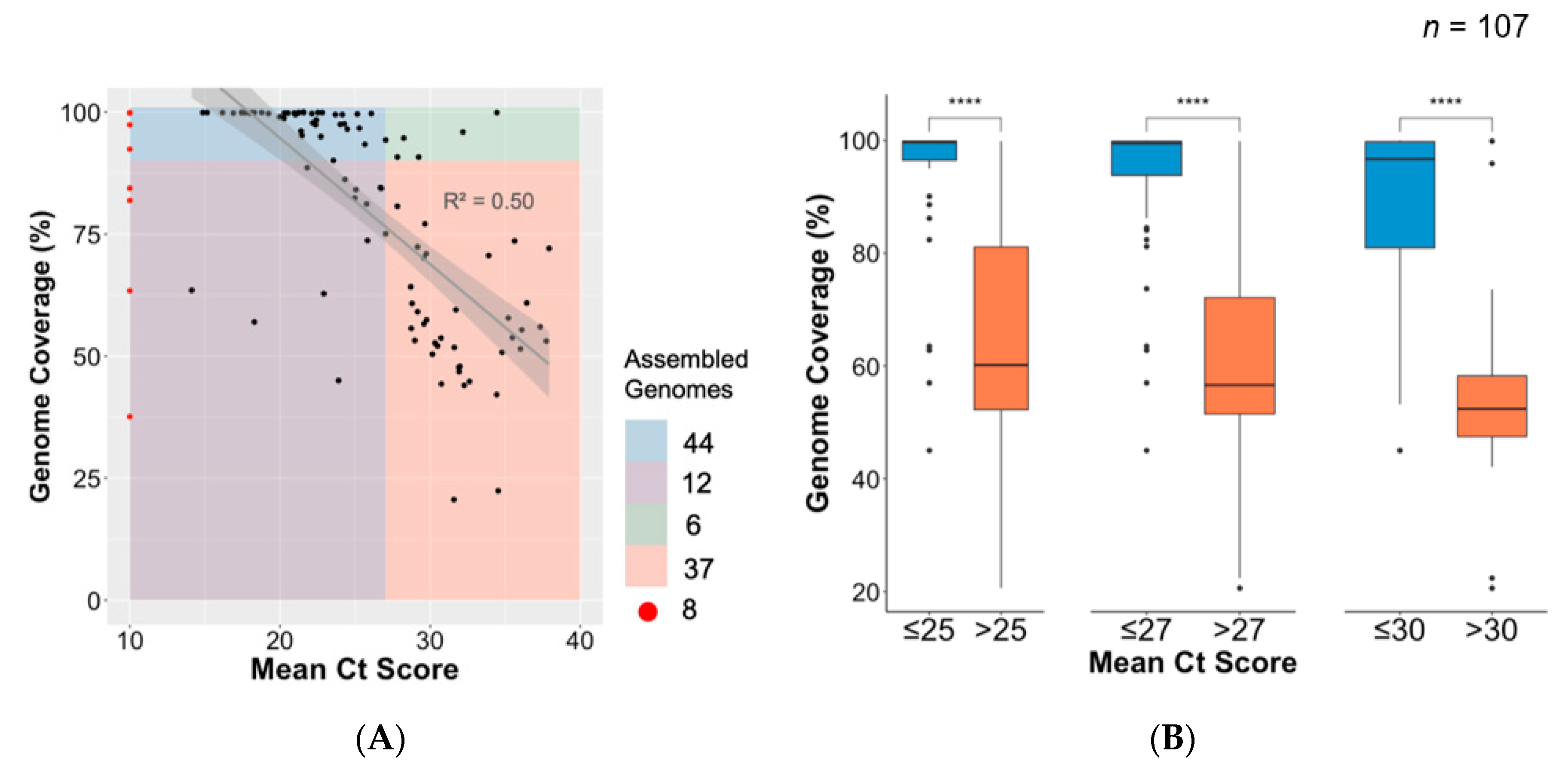
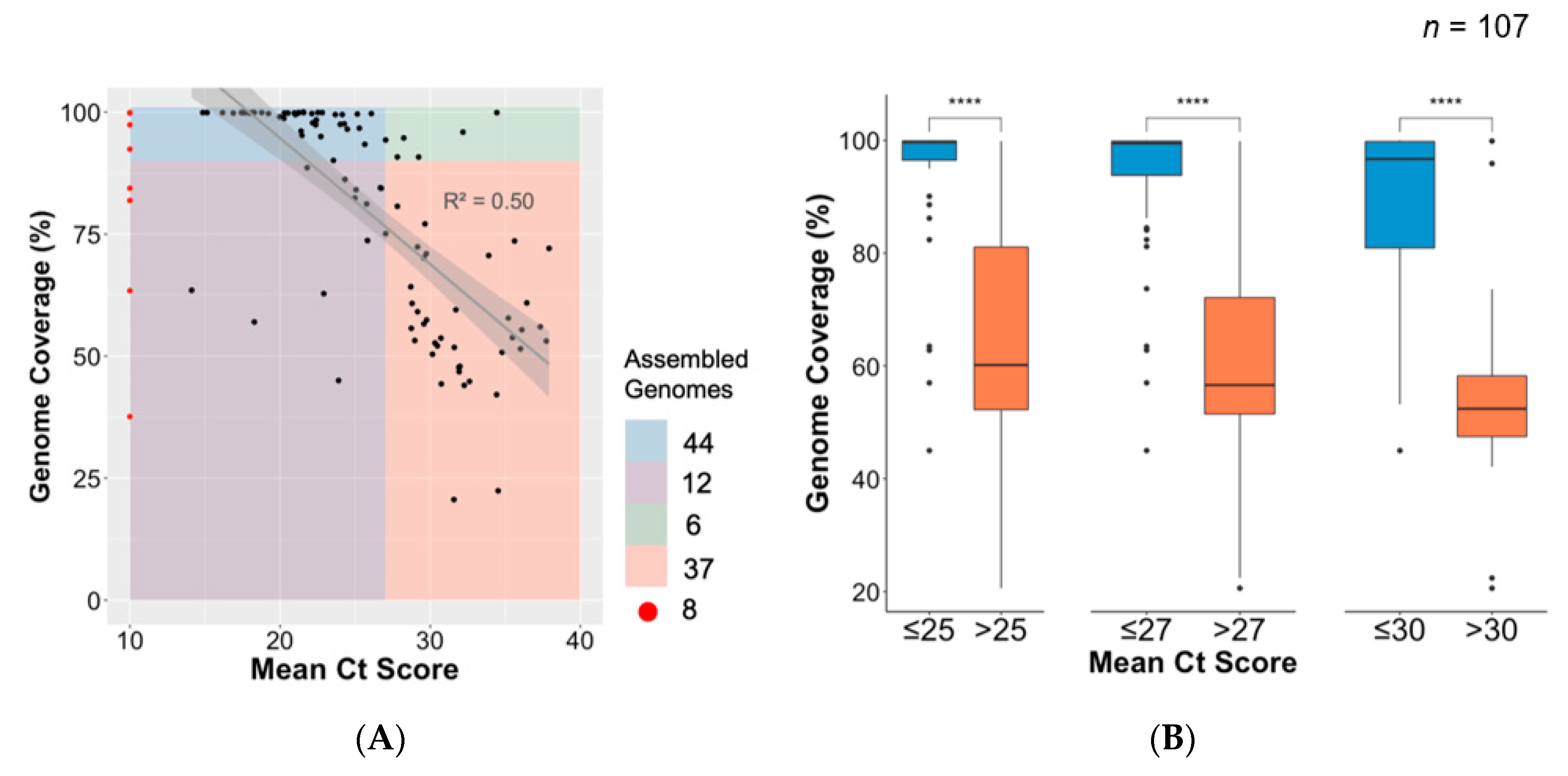
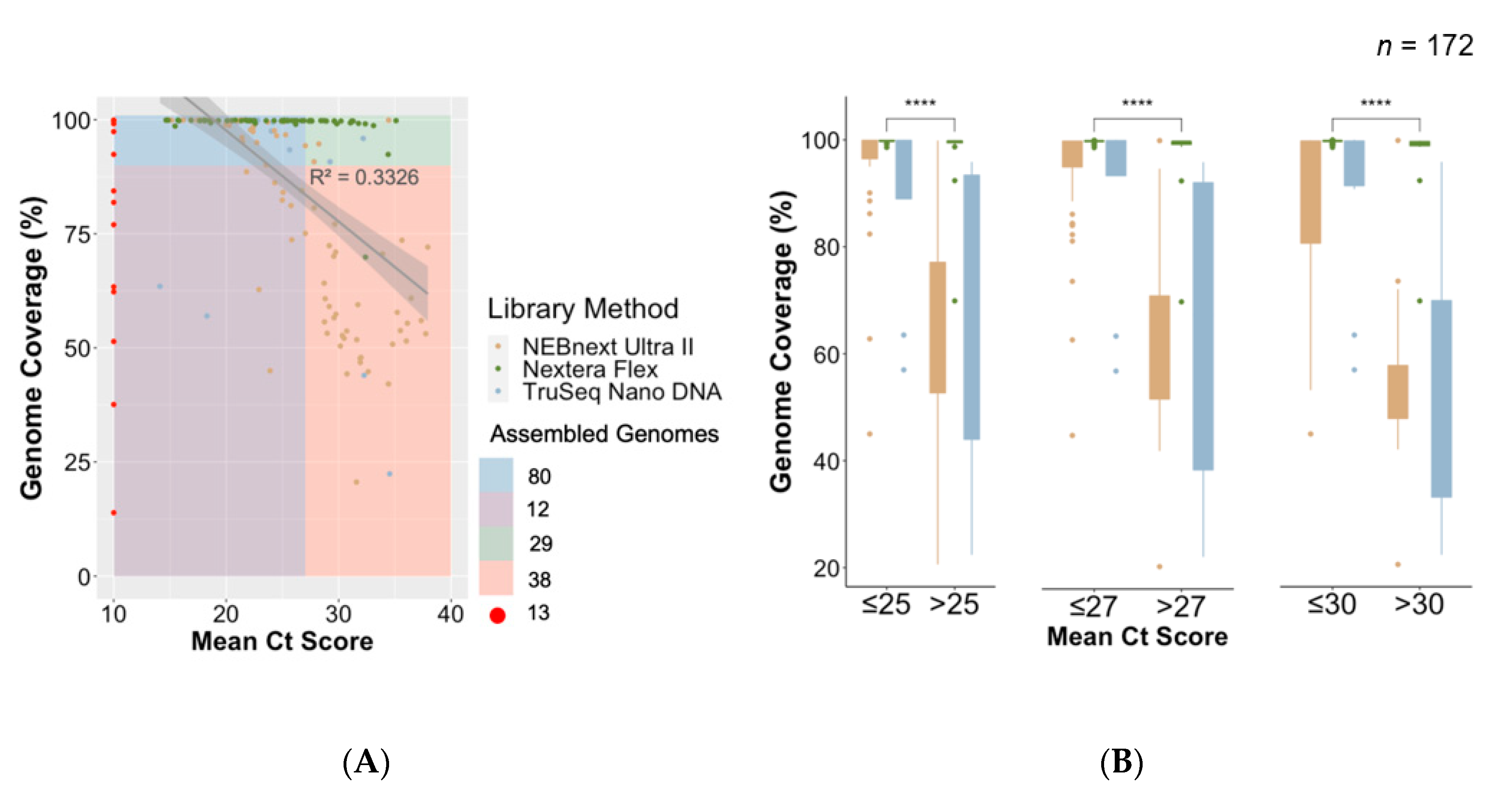
3.4. Association between Ct Value and Genome Coverage in an Outbreak
3.5. Comparison of Library Preparation Methods
3.6. Implementation of Nextera Flex and Improvement of Process and Success Rate of Sequencing
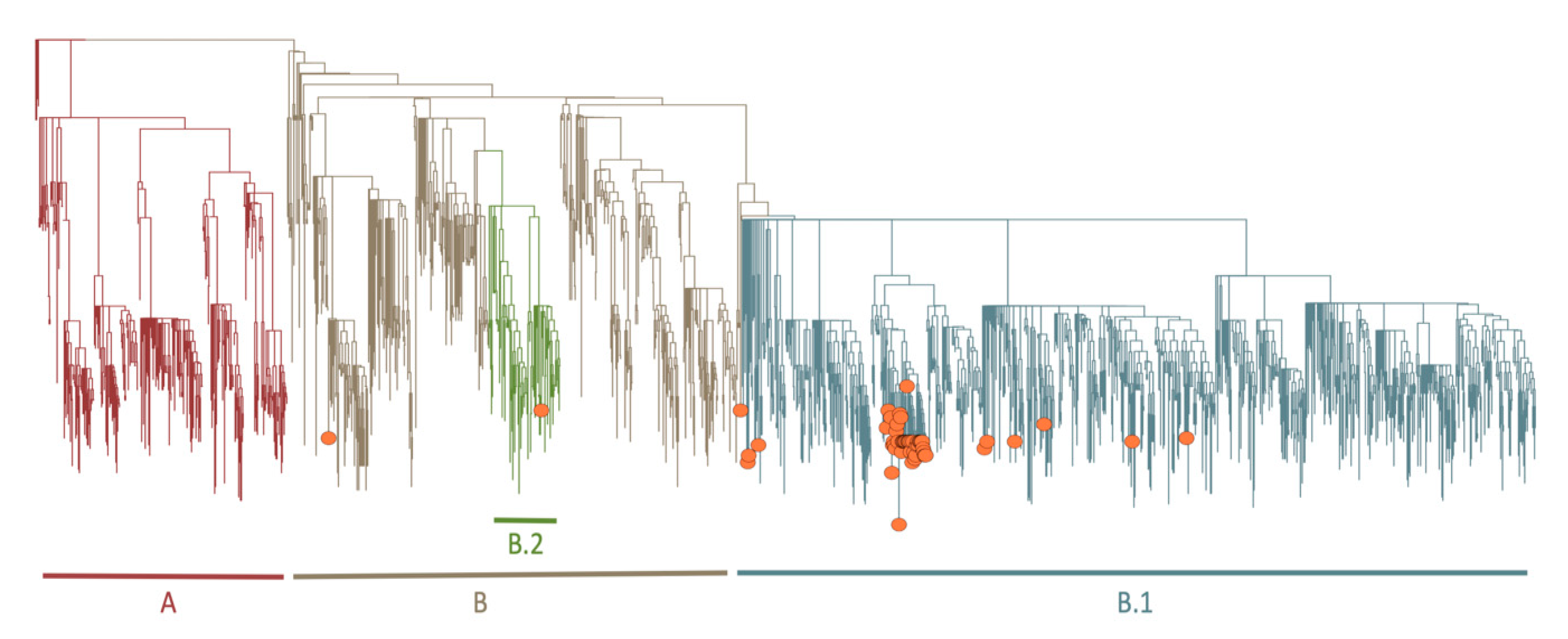
3.7. Phylogenetic and Lineage Analysis for Near Full-Length Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, S.; Zarbock, A. Coronaviruses and SARS-CoV-2: A Brief Overview. Anesth. Analg. 2020, 131, 93–96. [Google Scholar] [CrossRef]
- Maurier, F.; Beury, D.; Fléchon, L.; Varré, J.-S.; Touzet, H.; Goffard, A.; Hot, D.; Caboche, S. A complete protocol for whole-genome sequencing of virus from clinical samples: Application to coronavirus OC43. Virology 2019, 531, 141–148. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A.J.; Hewlett, E.L. Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Ladner, J.T.; Lemey, P.; Pybus, O.G.; Rambaut, A.; Holmes, E.C.; Andersen, K.G. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 2019, 4, 10–19. [Google Scholar] [CrossRef]
- Giandhari, J.; Pillay, S.; Wilkinson, E.; Tegally, H.; Sinayskiy, I.; Schuld, M.; Lourenço, J.; Chimukangara, B.; Lessells, R.J.; Moosa, Y.; et al. Early transmission of SARS-CoV-2 in South Africa: An epidemiological and phylogenetic report. MedRxiv 2020. [Google Scholar] [CrossRef]
- Deng, X.; Gu, W.; Federman, S.; du Plessis, L.; Pybus, O.G.; Faria, N.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.-Y.; et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 369, 582–587. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Helgason, A.; Jonsson, H.; Magnusson, O.T.; Melsted, P.; Norddahl, G.L.; Saemundsdottir, J.; Sigurdsson, A.; Sulem, P.; Agustsdottir, A.B.; et al. Spread of SARS-CoV-2 in the Icelandic Population. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Eden, J.-S.; Rockett, R.; Carter, I.; Rahman, H.; de Ligt, J.; Hadfield, J.; Storey, M.; Ren, X.; Tulloch, R.; Basile, K.; et al. An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. Virus Evol. 2020, 6, veaa027. [Google Scholar] [CrossRef]
- Gonzalez-Reiche, A.S.; Hernandez, M.M.; Sullivan, M.; Ciferri, B.; Alshammary, H.; Obla, A.; Fabre, S.; Kleiner, G.; Polanco, J.; Khan, Z.; et al. Introductions and early spread of SARS-CoV-2 in the New York City area. MedRxiv 2020. [Google Scholar] [CrossRef]
- Fauver, J.R.; Petrone, M.E.; Hodcroft, E.B.; Shioda, K.; Ehrlich, H.Y.; Watts, A.G.; Vogels, C.B.; Brito, A.F.; Alpert, T.; Muyombwe, A.; et al. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell Press 2020, 181, 990–996.e5. [Google Scholar] [CrossRef]
- Leung, K.S.-S.; Ng, T.T.-L.; Wu, A.K.-L.; Yau, M.C.-Y.; Lao, H.-Y.; Choi, M.-P.; Tam, K.K.-G.; Lee, L.-K.; Wong, B.K.-C.; Ho, A.Y.-M.; et al. A territory-wide study of early COVID-19 outbreak in Hong Kong community: A clinical, epidemiological and phylogenomic investigation. medRxiv 2020. [Google Scholar] [CrossRef]
- Quick, J. nCoV-2019 sequencing protocol V.1. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-bbmuik6w?version_warning=no (accessed on 10 June 2020). forked from Ebola virus sequencing protocol. [CrossRef] [Green Version]
- Cleemput, S.; Dumon, W.; Fonseca, V.; Abdool Karim, W.; Giovanetti, M.; Carlos Alcantara, L.; Deforche, K.; de Oliveira, T. Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics 2020, 36, 3552–3555. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Carlos Alcantara, L.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Holmes, E.C.; Hill, V.; OToole, A.; McCrone, J.; Ruis, C.; du Plessis, L.; Pybus, O. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. BioRxiv 2020. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavaré, S.; Miura, R.M. Some mathematical questions in biology: DNA sequence analysis lectures on mathematics in the life sciences. Stat. Med. 1986, 4, 523–524. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Boil. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinforma. 2020, 69, e96. [Google Scholar] [CrossRef]
- Lessells, R.; Moosa, Y.; De Oliveira, T. Report into a nosocomial outbreak of coronavirus disease 2019 (COVID-19) at Netcare St. Augustine’s Hospital. Available online: https://www.krisp.org.za/news.php?id=421 (accessed on 22 June 2020).
- Seth-Smith, H.M.B.; Bonfiglio, F.; Cuénod, A.; Reist, J.; Egli, A.; Wüthrich, D. Evaluation of rapid library preparation protocols for whole genome sequencing based outbreak investigation. Front. Public Heal. 2019, 7, 241. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coverage (% of SARS-CoV-2 Genome) | ||||
|---|---|---|---|---|
| Sequence | TruSeq DNA Nano | NEBnext Ultra II | Nextera Flex | Ct Value |
| KRISP_0002 | 97.5 | 98.3 | 98.5 | 24.0 |
| KRISP_0004 | 99.5 | 98.3 | 98.1 | 24.1 |
| KRISP_0019 | 97.4 | 89.9 | 90.2 | NA |
| KRISP_0021 | 63.5 | 99.1 | 82.7 | 14.1 |
| KRISP_0024 | - | 95.2 | 94.6 | 21.5 |
| KRISP_0026 | - | 99.8 | 99.9 | 17.9 |
| KRISP_0028 | - | 96.1 | 98.1 | 21.4 |
| KRISP_0031 | - | 86.2 | 84.1 | 24.3 |
| KRISP_016 | 99.8 | - | 99.2 | NA |
| KRISP_017 | 99.9 | - | 99.9 | NA |
| KRISP_006 | 99.5 | - | 96.3 | 21.0 |
| KRISP_007 | 99.9 | - | 99.6 | 25.5 |
| KRISP_003 | 25.3 | 92.4 | - | Undetermined |
| KRISP_010 | 93.4 | 92.3 | - | 25.6 |
| KRISP_014 | 81.9 | 70.6 | - | NA |
| KRISP_013 | 63.4 | 73.0 | - | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pillay, S.; Giandhari, J.; Tegally, H.; Wilkinson, E.; Chimukangara, B.; Lessells, R.; Moosa, Y.; Mattison, S.; Gazy, I.; Fish, M.; et al. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. Genes 2020, 11, 949. https://doi.org/10.3390/genes11080949
Pillay S, Giandhari J, Tegally H, Wilkinson E, Chimukangara B, Lessells R, Moosa Y, Mattison S, Gazy I, Fish M, et al. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. Genes. 2020; 11(8):949. https://doi.org/10.3390/genes11080949
Chicago/Turabian StylePillay, Sureshnee, Jennifer Giandhari, Houriiyah Tegally, Eduan Wilkinson, Benjamin Chimukangara, Richard Lessells, Yunus Moosa, Stacey Mattison, Inbal Gazy, Maryam Fish, and et al. 2020. "Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic" Genes 11, no. 8: 949. https://doi.org/10.3390/genes11080949
APA StylePillay, S., Giandhari, J., Tegally, H., Wilkinson, E., Chimukangara, B., Lessells, R., Moosa, Y., Mattison, S., Gazy, I., Fish, M., Singh, L., Khanyile, K. S., San, J. E., Fonseca, V., Giovanetti, M., Alcantara, L. C., Jr., & de Oliveira, T. (2020). Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. Genes, 11(8), 949. https://doi.org/10.3390/genes11080949