Leigh Syndrome in a Pedigree Harboring the m.1555A>G Mutation in the Mitochondrial 12S rRNA

 , , and
, , and 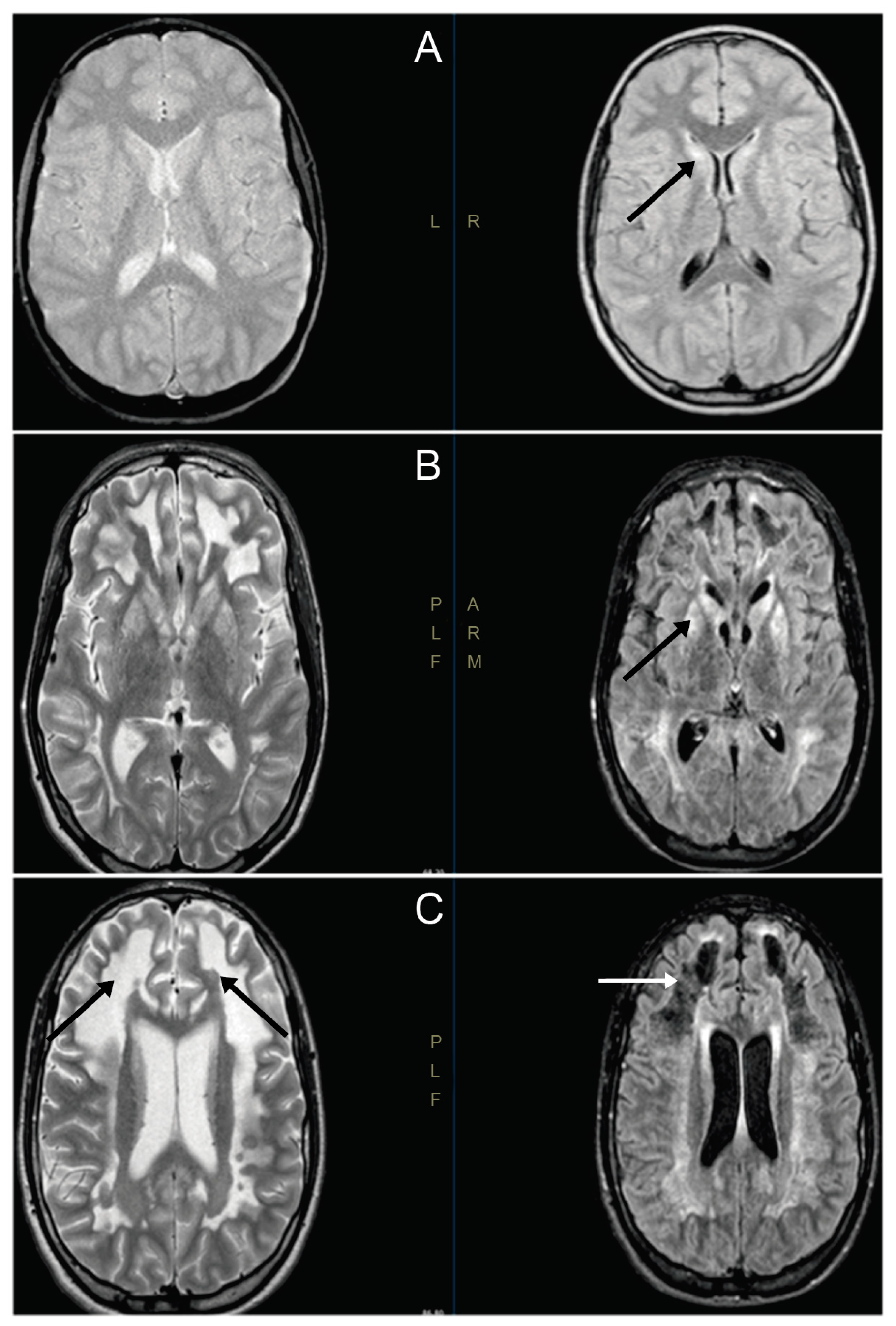{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Issues
2.2. Biochemical Studies
2.3. DNA Studies
2.4. Cybrids
2.5. Statistical Analysis
3. Results
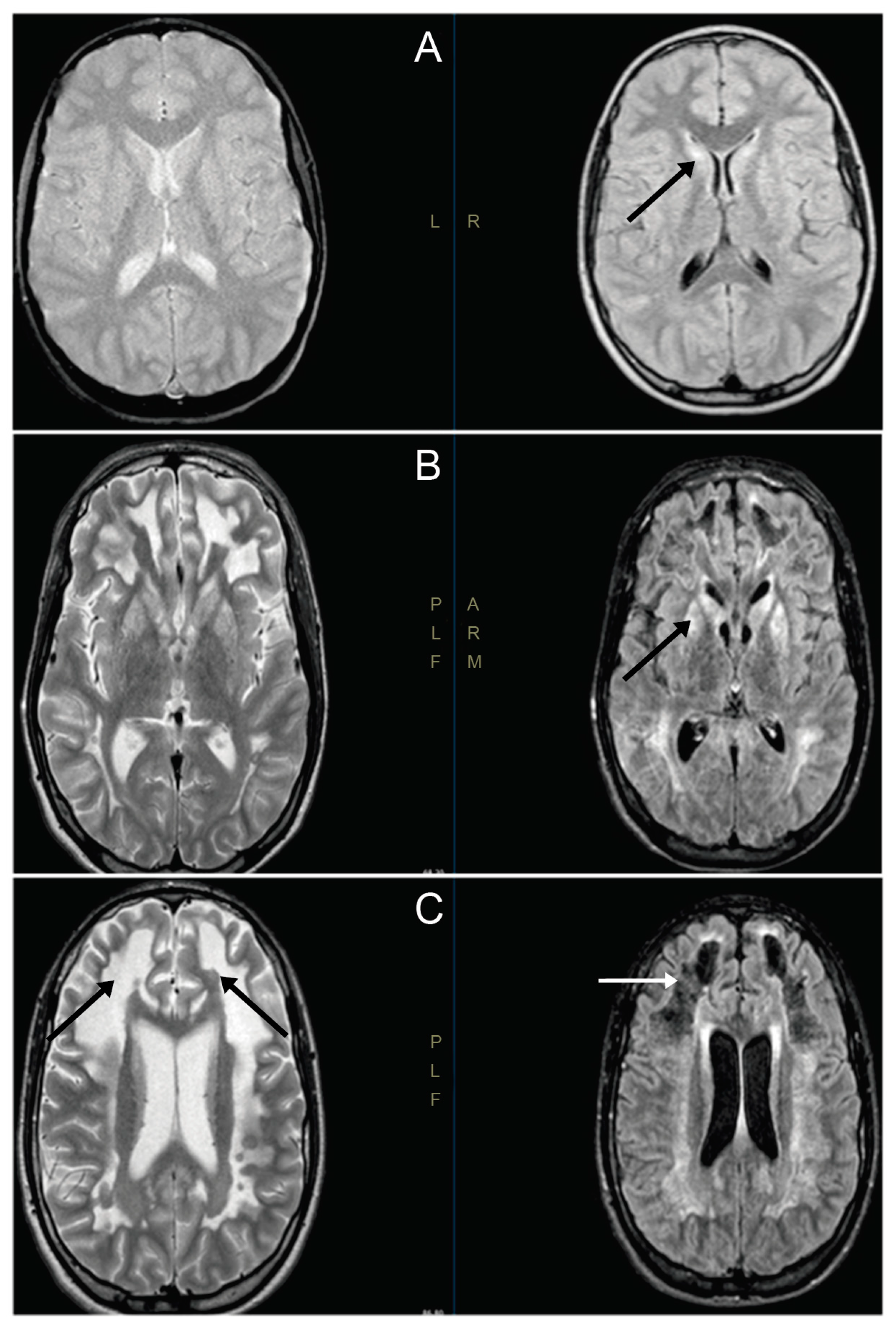
3.1. Clinical Cases
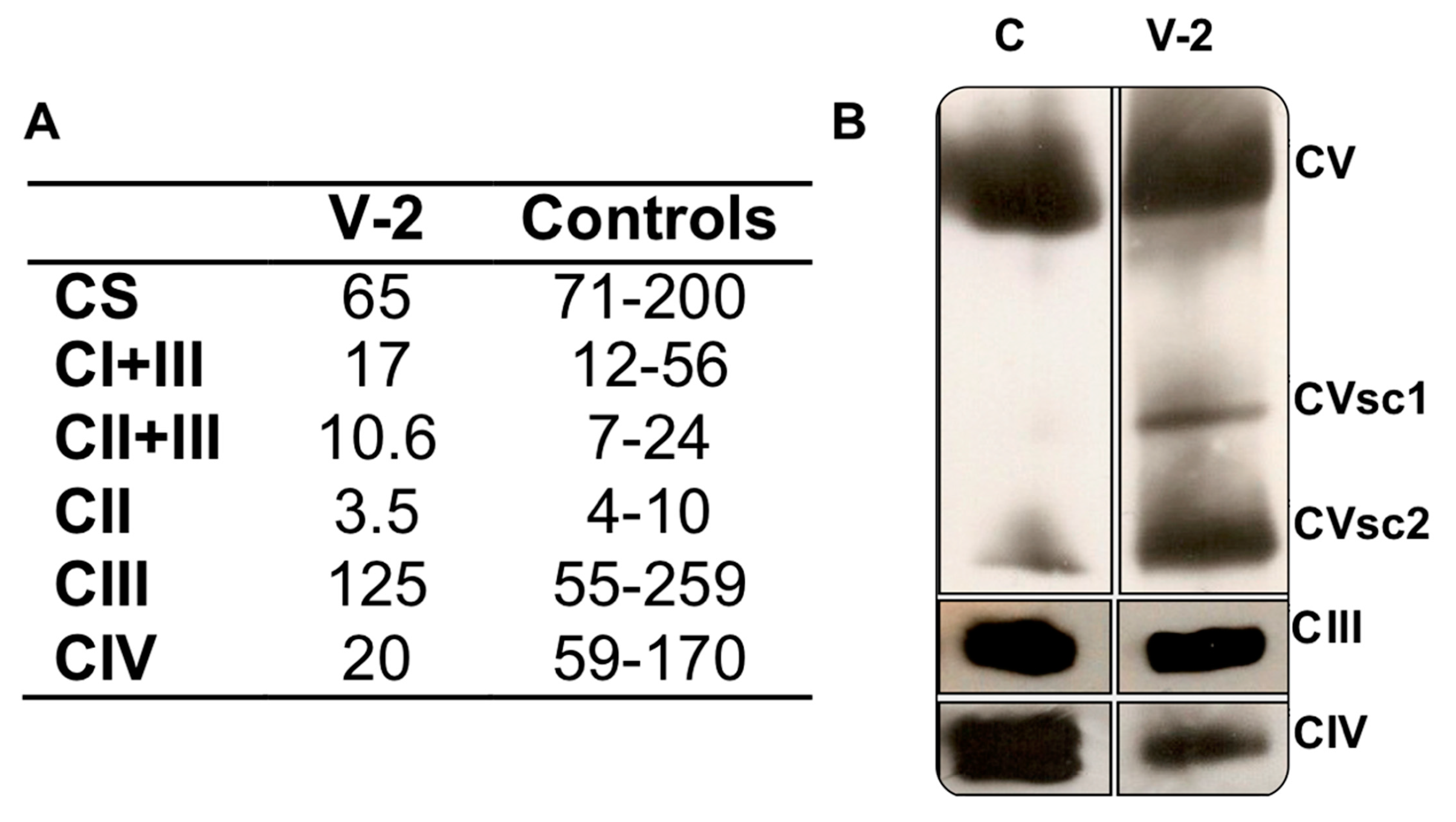
3.2. Respiratory Complex Assays
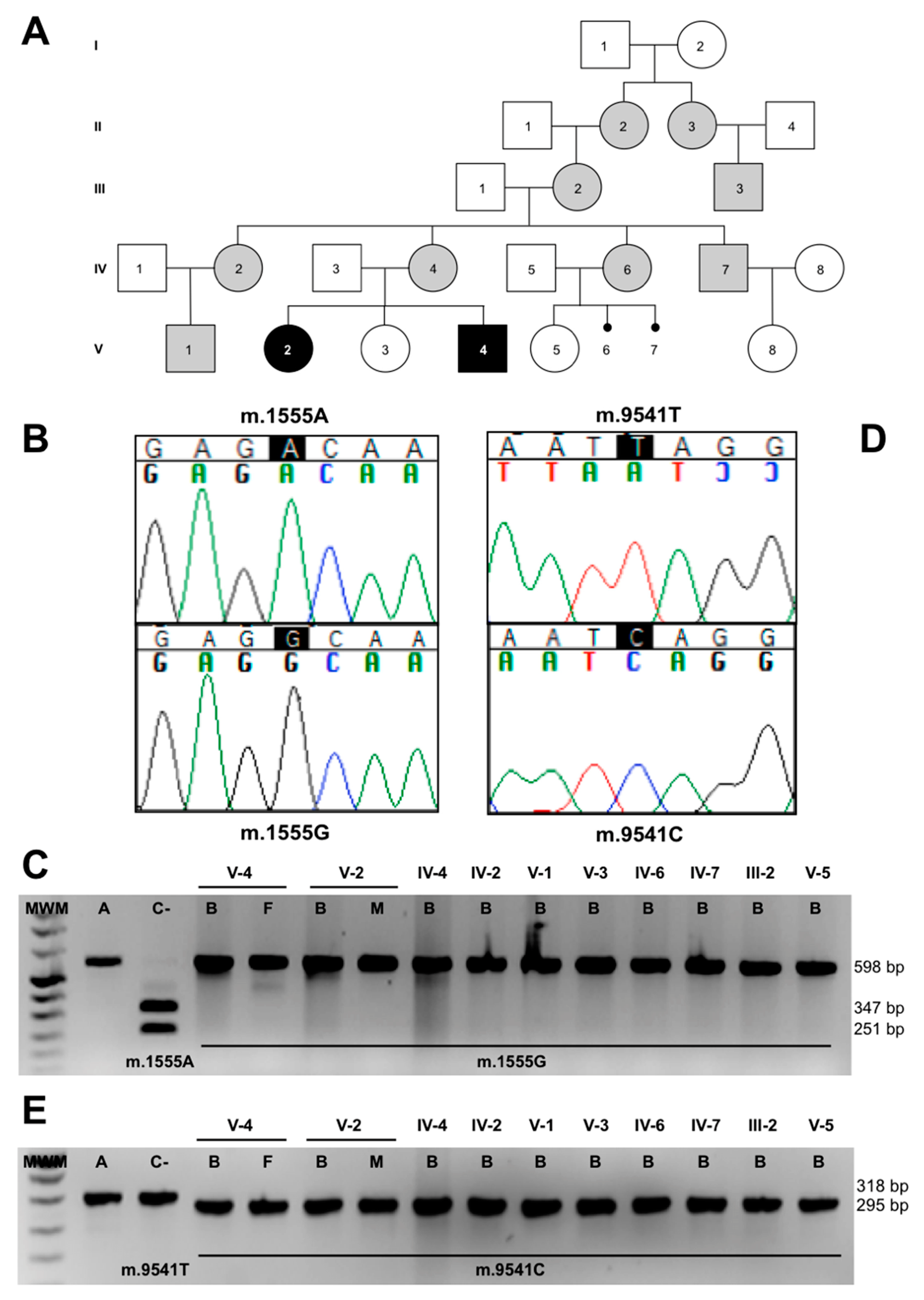
3.3. Molecular-Genetic Analysis
3.4. Cybrids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.H.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2013, 85, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.; López-Gallardo, E.; Díez-Sánchez, C.; López-Pérez, M.J.; Ruiz-Pesini, E. 20 years of human mtDNA pathologic point mutations: Carefully reading the pathogenicity criteria. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2009, 1787, 476–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, D.R.; Rahman, J.; Rahman, S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2015, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Öztas, S.; Qiu, W.-Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic–induced and non–syndromic deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, R.; Wang, Q.; Yan, Q.; Deng, J.-H.; Han, N.; Bai, Y.; Young, W.-Y.; Guan, M.-X. Maternally Inherited Aminoglycoside-Induced and Nonsyndromic Deafness Is Associated with the Novel C1494T Mutation in the Mitochondrial 12S rRNA Gene in a Large Chinese Family. Am. J. Hum. Genet. 2003, 74, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gérard, B.; Rötig, A.; Saudubray, J.; Munnich, A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 1994, 228, 35–51. [Google Scholar] [CrossRef]
- Herrero-Martín, M.D.; Pineda, M.; Briones, P.; López-Gallardo, E.; Carreras, M.; Benac, M.; Idoate, M.A.; Vilaseca, M.A.; Artuch, R.; López-Pérez, M.J.; et al. A new pathologic mitochondrial DNA mutation in the cytochrome oxidase subunit I (MT-CO1). Hum. Mutat. 2008, 29, E112–E122. [Google Scholar] [CrossRef]
- Andreu, A.L.; Martínez, R.; Martí, R.; Garcia-Arumi, E. Quantification of mitochondrial DNA copy number: Pre-analytical factors. Mitochondrion 2009, 9, 242–246. [Google Scholar] [CrossRef]
- Grau, D.P.; Pérez-Delgado, L.; Gómez-Díaz, C.; Fraile-Rodrigo, J.; Montoya, J.; Ruiz-Pesini, E. Mitochondrial ribosome and Ménière’s disease: A pilot study. Eur. Arch. Oto-Rhino-Laryngol. 2012, 269, 2003–2008. [Google Scholar] [CrossRef]
- Emperador, S.; Vidal, M.; Hernandez-Ainsa, C.; Ruiz-Ruiz, C.; Woods, D.; Morales-Becerra, A.; Arruga, J.; Artuch, R.; López-Gallardo, E.; Bayona-Bafaluy, M.P.; et al. The Decrease in Mitochondrial DNA Mutation Load Parallels Visual Recovery in a Leber Hereditary Optic Neuropathy Patient. Front. Mol. Neurosci. 2018, 12, 61. [Google Scholar] [CrossRef]
- Wittig, I.; Carrozzo, R.; Santorelli, F.M.; Schägger, H. Functional assays in high-resolution clear native gels to quantify mitochondrial complexes in human biopsies and cell lines. Electrophoresis 2007, 28, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- López-Gallardo, E.; Solano, A.; Herrero-Martín, M.D.; Martínez-Romero, Í.; Castaño-Pérez, M.D.; Andreu, A.L.; Herrera, A.; López-Pérez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. J. Med. Genet. 2008, 46, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Duran, A.; Grau, D.P.; López-Gallardo, E.; Díez-Sánchez, C.; Montoya, J.; López-Pérez, M.J.; Ruiz-Pesini, E. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum. Mol. Genet. 2010, 19, 3343–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Gallardo, E.; Emperador, S.; Solano, A.; Llobet, L.; Martín-Navarro, A.; López-Pérez, M.J.; Briones, P.; Pineda, M.; Artuch, R.; Barraquer, E.; et al. Expanding the clinical phenotypes of MT-ATP6 mutations. Hum. Mol. Genet. 2014, 23, 6191–6200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- MITOMAP: A Human Mitochondrial Genome Database. Available online: http://www.mitomap.org (accessed on 19 June 2020).
- Pereira, L.; Soares, P.; Radivojac, P.; Li, B.; Samuels, D.C. Comparing Phylogeny and the Predicted Pathogenicity of Protein Variations Reveals Equal Purifying Selection across the Global Human mtDNA Diversity. Am. J. Hum. Genet. 2011, 88, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Martín-Navarro, A.; Gaudioso-Simón, A.; Alvarez-Jarreta, J.; Montoya, J.; Mayordomo, E.; Ruiz-Pesini, E. Machine learning classifier for identification of damaging missense mutations exclusive to human mitochondrial DNA-encoded polypeptides. BMC Bioinform. 2017, 18, 158. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; He, Z.; Tang, X.; Zheng, J.; Jin, X.; Zhu, Y.; Ren, X.; Zhou, M.; Wang, M.; Gong, S.; et al. Contribution of the tRNAIle 4317A→G mutation to the phenotypic manifestation of the deafness-associated mitochondrial 12S rRNA 1555A→G mutation. J. Boil. Chem. 2018, 293, 3321–3334. [Google Scholar] [CrossRef] [Green Version]
- Yamasoba, T.; Goto, Y.-I.; Oka, Y.; Nishino, I.; Tsukuda, K.; Nonaka, I. Atypical muscle pathology and a survey of cis-mutations in deaf patients harboring a 1555 A-to-G point mutation in the mitochondrial ribosomal RNA gene. Neuromuscul. Disord. 2002, 12, 506–512. [Google Scholar] [CrossRef]
- Kouzaki, H.; Suzuki, M.; Shimizu, T. Immunohistochemical and ultrastructural abnormalities in muscle from a patient with sensorineural hearing loss related to a 1555 A-to-G mitochondrial mutation. J. Clin. Neurosci. 2007, 14, 603–607. [Google Scholar] [CrossRef]
- Nye, J.S.; Hayes, E.A.; Amendola, M.; Vaughn, D.; Charrow, J.; McLone, D.G.; Speer, M.C.; Nance, W.E.; Pandya, A. Myelocystocele-cloacal exstrophy in a pedigree with a mitochondrial 12S rRNA mutation, aminoglycoside-induced deafness, pigmentary disturbances, and spinal anomalies. Teratology 2000, 61, 165–171. [Google Scholar] [CrossRef]
- Chen, H.; Zheng, J.; Xue, L.; Meng, Y.; Wang, Y.; Zheng, B.; Fang, F.; Shi, S.; Qiu, Q.; Jiang, P.; et al. The 12S rRNA A1555G mutation in the mitochondrial haplogroup D5a is responsible for maternally inherited hypertension and hearing loss in two Chinese pedigrees. Eur. J. Hum. Genet. 2012, 20, 607–612. [Google Scholar] [CrossRef]
- Mezghani, N.; Mnif, M.; Mkaouar-Rebai, E.; Kallel, N.; Charfi, N.; Abid, M.; Fakhfakh, F. A maternally inherited diabetes and deafness patient with the 12S rRNA m.1555A>G and the ND1 m.3308T>C mutations associated with multiple mitochondrial deletions. Biochem. Biophys. Res. Commun. 2013, 431, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Skou, A.-S.; Tranebjærg, L.; Jensen, T.; Hasle, H. Mitochondrial 12S Ribosomal RNA A1555G Mutation Associated with Cardiomyopathy and Hearing Loss following High-Dose Chemotherapy and Repeated Aminoglycoside Exposure. J. Pediatr. 2014, 164, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Alila-Fersi, O.; Chamkha, I.; Majdoub, I.; Gargouri, L.; Mkaouar-Rebai, E.; Tabebi, M.; Tlili, A.; Keskes, L.; Mahfoudh, A.; Fakhfakh, F. Co segregation of the m.1555A>G mutation in the MT-RNR1 gene and mutations in MT-ATP6 gene in a family with dilated mitochondrial cardiomyopathy and hearing loss: A whole mitochondrial genome screening. Biochem. Biophys. Res. Commun. 2017, 484, 71–78. [Google Scholar] [CrossRef]
- Santorelli, F.M.; Tanji, K.; Manta, P.; Casali, C.; Krishna, S.; Hays, A.P.; Mancini, N.M.; DiMauro, S.; Hirano, M. Maternally inherited cardiomyopathy: An atypical presentation of the mtDNA 12S rRNA gene A1555G mutation. Am. J. Hum. Genet. 1999, 64, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Campos, Y.; García, A.; Lopez, A.; Jiménez, S.; Rubio, J.; Del Hoyo, P.; Bustos, F.; Martín, M.; Cabello, A.; Ricoy, J.; et al. Cosegregation of the mitochondrial DNA A1555G and G4309A mutations results in deafness and mitochondrial myopathy. Muscle Nerve 2002, 25, 185–188. [Google Scholar] [CrossRef]
- Czajkowski, A.; Mounier, A.; Delacroix, L.; Malgrange, B. Pluripotent stem cell-derived cochlear cells: A challenge in constant progress. Cell. Mol. Life Sci. 2018, 76, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Deguine, O.; Chappert, C.; Fonta, C.; Imbert, M. Prenatal lesioning of vestibular organ by aminoglycosides. NeuroReport 1996, 7, 2435–2438. [Google Scholar] [CrossRef]
- Mantovani, A.; Calamandrei, G. Delayed developmental effects following prenatal exposure to drugs. Curr. Pharm. Des. 2001, 7, 859–880. [Google Scholar] [CrossRef]
- Emperador, S.; Grau, D.P.; Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Martín-Navarro, A.; López-Pérez, M.J.; Montoya, J.; Ruiz-Pesini, E. An MRPS12 mutation modifies aminoglycoside sensitivity caused by 12S rRNA mutations. Front. Genet. 2015, 5, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potluri, P.; Davila, A.; Ruiz-Pesini, E.; Mishmar, D.; O’Hearn, S.; Hancock, S.; Simon, M.; Scheffler, I.E.; Wallace, D.C.; Procaccio, V. A novel NDUFA1 mutation leads to a progressive mitochondrial complex I-specific neurodegenerative disease. Mol. Genet. Metab. 2009, 96, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habbane, M.; Llobet, L.; Bayona-Bafaluy, M.P.; Bárcena, J.E.; Ceberio, L.; Gómez-Díaz, C.; Gort, L.; Artuch, R.; Montoya, J.; Ruiz-Pesini, E. Leigh Syndrome in a Pedigree Harboring the m.1555A>G Mutation in the Mitochondrial 12S rRNA. Genes 2020, 11, 1007. https://doi.org/10.3390/genes11091007
Habbane M, Llobet L, Bayona-Bafaluy MP, Bárcena JE, Ceberio L, Gómez-Díaz C, Gort L, Artuch R, Montoya J, Ruiz-Pesini E. Leigh Syndrome in a Pedigree Harboring the m.1555A>G Mutation in the Mitochondrial 12S rRNA. Genes. 2020; 11(9):1007. https://doi.org/10.3390/genes11091007
Chicago/Turabian StyleHabbane, Mouna, Laura Llobet, M. Pilar Bayona-Bafaluy, José E. Bárcena, Leticia Ceberio, Covadonga Gómez-Díaz, Laura Gort, Rafael Artuch, Julio Montoya, and Eduardo Ruiz-Pesini. 2020. "Leigh Syndrome in a Pedigree Harboring the m.1555A>G Mutation in the Mitochondrial 12S rRNA" Genes 11, no. 9: 1007. https://doi.org/10.3390/genes11091007