Aneuploidy and DNA Methylation as Mirrored Features of Early Human Embryo Development


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Incidence of Aneuploidy in Ontogenesis
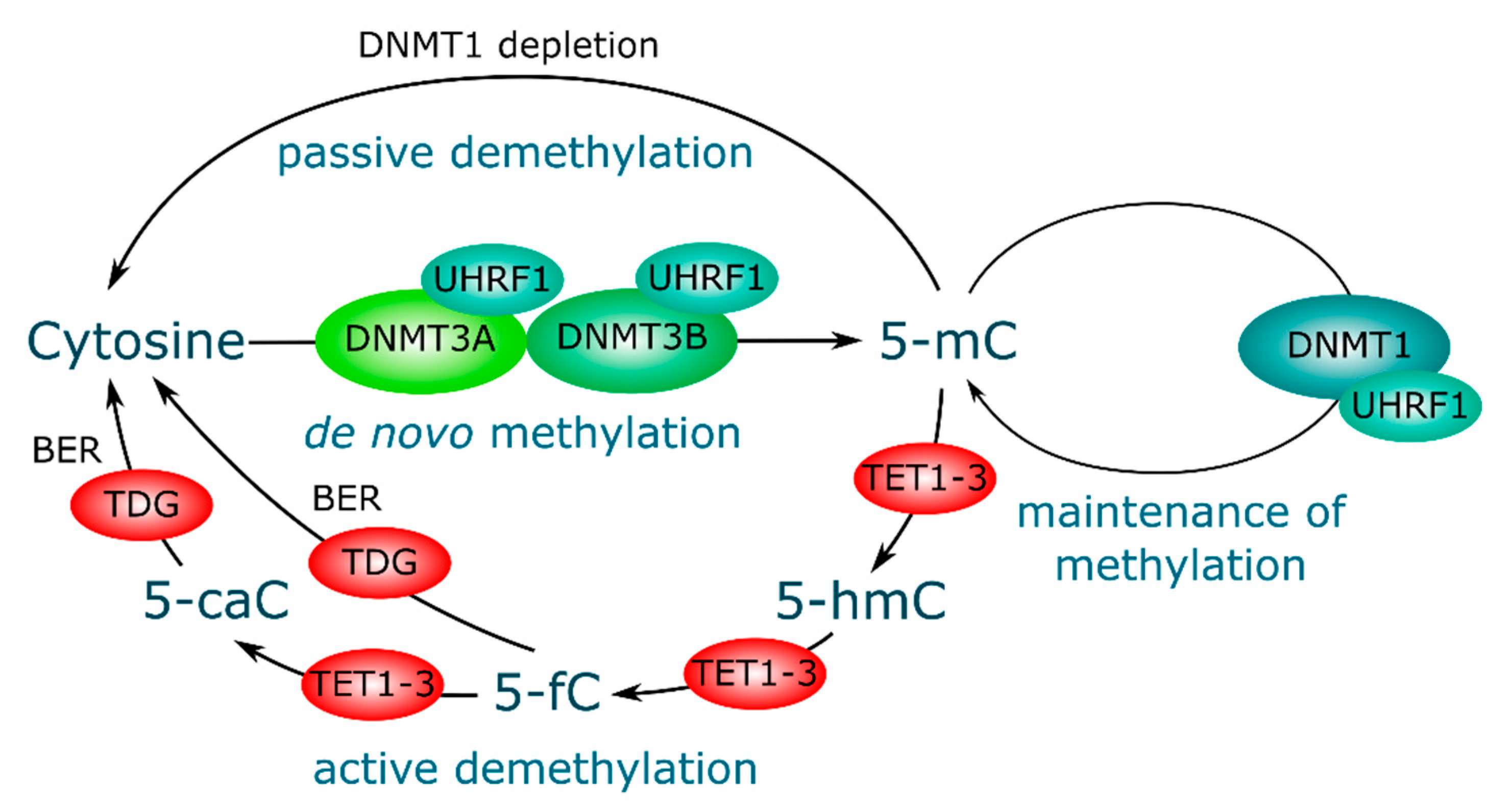
3. DNA Methylation and Demethylation, Mechanisms and Key Players
4. DNA Methylation and Aneuploidy in Gametogenesis
5. DNA Methylation and Aneuploidy at the Cleavage Stage
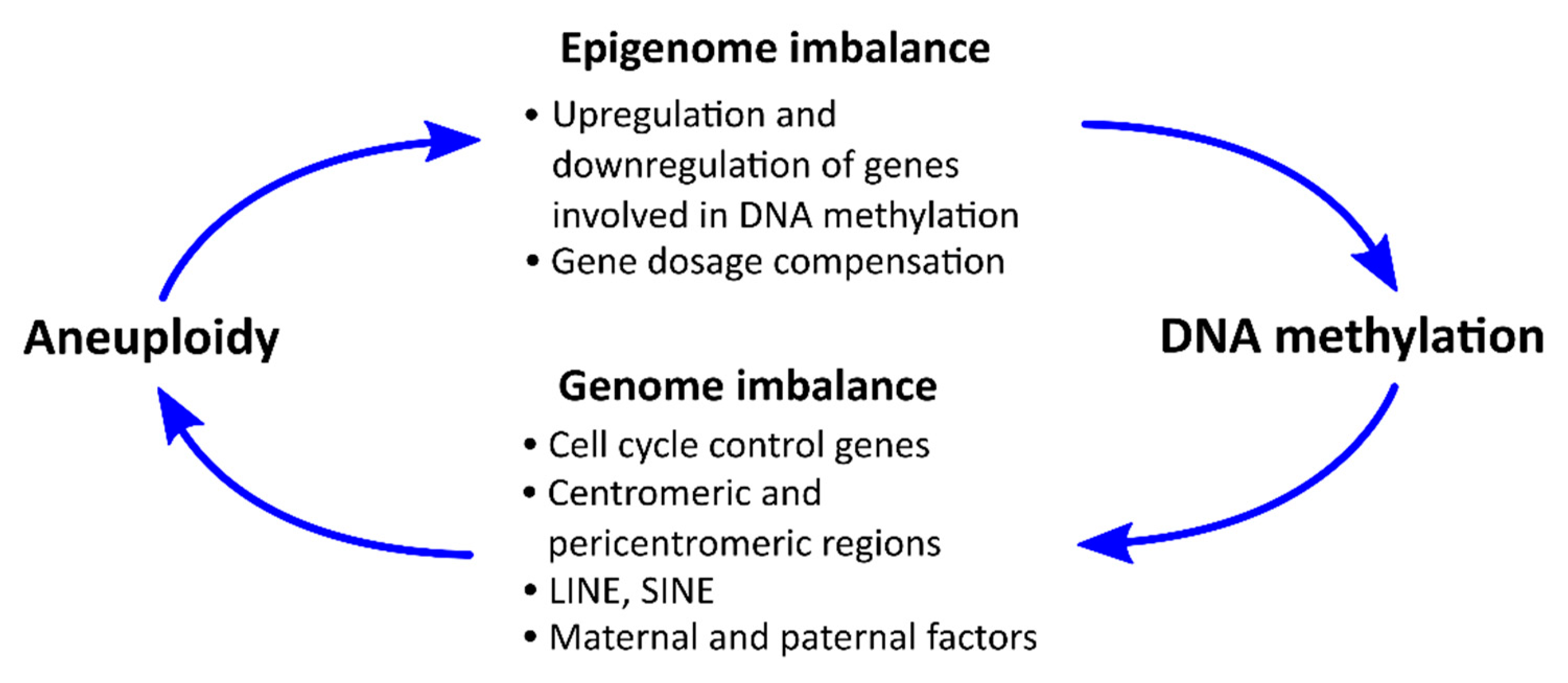
6. Potential Mechanisms for the Relationship between DNA Methylation and Aneuploidy
6.1. Influence of Aneuploidy on Certain Chromosomes on the Level of DNA Methylation
6.2. Effects of DNA Methylation on the Incidence of Aneuploidy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nikitina, T.V.; Sazhenova, E.A.; Tolmacheva, E.N.; Sukhanova, N.N.; Kashevarova, A.A.; Skryabin, N.A.; Vasilyev, S.A.; Nemtseva, T.N.; Yuriev, S.Y.; Lebedev, I.N. Comparative cytogenetic analysis of spontaneous abortions in recurrent and sporadic pregnancy losses. Biomed. Hub 2016, 1, 446099. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Alfarawati, S.; Spath, K.; Jaroudi, S.; Sarasa, J.; Enciso, M.; Wells, D. The origin and impact of embryonic aneuploidy. Hum. Genet. 2013, 132, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Wells, D. Aneuploidy in the human blastocyst. Cytogenet. Genome Res. 2011, 133, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Ghevaria, H.; SenGupta, S.; Shmitova, N.; Serhal, P.; Delhanty, J. The origin and significance of additional aneuploidy events in couples undergoing preimplantation genetic diagnosis for translocations by array comparative genomic hybridization. Reprod. Biomed. Online 2016, 32, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Hassold, T.; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet. 2001, 2, 280–291. [Google Scholar] [CrossRef]
- Dupont, C.; Segars, J.; DeCherney, A.; Bavister, B.D.; Armant, D.R.; Brenner, C.A. Incidence of chromosomal mosaicism in morphologically normal nonhuman primate preimplantation embryos. Fertil. Steril. 2010, 93, 2545–2550. [Google Scholar] [CrossRef] [Green Version]
- Tsuiko, O.; Catteeuw, M.; Esteki, M.Z.; Destouni, A.; Pascottini, O.B.; Besenfelder, U.; Havlicek, V.; Smits, K.; Kurg, A.; Salumets, A.; et al. Genome stability of bovine in vivo-conceived cleavage-stage embryos is higher compared to in vitro-produced embryos. Hum. Reprod. 2017, 32, 2348–2357. [Google Scholar] [CrossRef]
- Lightfoot, D.A.; Kouznetsova, A.; Mahdy, E.; Wilbertz, J.; Hoog, C. The fate of mosaic aneuploid embryos during mouse development. Dev. Biol. 2006, 289, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Chen, T. DNA methylation reprogramming during mammalian development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Veronese, A. Genome DNA methylation, aneuploidy and immunity in cancer. Epigenomics 2020. [Google Scholar] [CrossRef]
- Herrera, L.A.; Prada, D.; Andonegui, M.A.; Duenas-Gonzalez, A. The epigenetic origin of aneuploidy. Curr. Genom. 2008, 9, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Delhanty, J.D.; SenGupta, S.B.; Ghevaria, H. How common is germinal mosaicism that leads to premeiotic aneuploidy in the female? J. Assist. Reprod. Genet. 2019, 36, 2403–2418. [Google Scholar] [CrossRef] [Green Version]
- Gruhn, J.R.; Zielinska, A.P.; Shukla, V.; Blanshard, R.; Capalbo, A.; Cimadomo, D.; Nikiforov, D.; Chan, A.C.; Newnham, L.J.; Vogel, I.; et al. Chromosome errors in human eggs shape natural fertility over reproductive life span. Science 2019, 365, 1466–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Ma, P.; Zhu, W.; Schultz, R.M. Age-associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev. Biol. 2008, 316, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Luciano, A.M.; Franciosi, F.; Lodde, V.; Tessaro, I.; Corbani, D.; Modina, S.C.; Peluso, J.J. Oocytes isolated from dairy cows with reduced ovarian reserve have a high frequency of aneuploidy and alterations in the localization of progesterone receptor membrane component 1 and aurora kinase B. Biol. Reprod. 2013, 88, 58. [Google Scholar] [CrossRef]
- Garcia-Mengual, E.; Trivino, J.C.; Saez-Cuevas, A.; Bataller, J.; Ruiz-Jorro, M.; Vendrell, X. Male infertility: Establishing sperm aneuploidy thresholds in the laboratory. J. Assist. Reprod. Genet. 2019, 36, 371–381. [Google Scholar] [CrossRef]
- Templado, C.; Vidal, F.; Estop, A. Aneuploidy in human spermatozoa. Cytogenet. Genome Res. 2011, 133, 91–99. [Google Scholar] [CrossRef]
- Petit, F.M.; Frydman, N.; Benkhalifa, M.; Le Du, A.; Aboura, A.; Fanchin, R.; Frydman, R.; Tachdjian, G. Could sperm aneuploidy rate determination be used as a predictive test before intracytoplasmic sperm injection? J. Androl. 2005, 26, 235–241. [Google Scholar] [CrossRef]
- Hassold, T.; Pettay, D.; Robinson, A.; Uchida, I. Molecular studies of parental origin and mosaicism in 45,X conceptuses. Hum. Genet. 1992, 89, 647–652. [Google Scholar] [CrossRef]
- Viotti, M. Preimplantation genetic testing for chromosomal abnormalities: Aneuploidy, mosaicism, and structural rearrangements. Genes 2020, 11, 602. [Google Scholar] [CrossRef]
- Ruangvutilert, P.; Delhanty, J.D.; Rodeck, C.H.; Harper, J.C. Relative efficiency of FISH on metaphase and interphase nuclei from non-mosaic trisomic or triploid fibroblast cultures. Prenat. Diagn. 2000, 20, 159–162. [Google Scholar] [CrossRef]
- Coonen, E.; Derhaag, J.G.; Dumoulin, J.C.; van Wissen, L.C.; Bras, M.; Janssen, M.; Evers, J.L.; Geraedts, J.P. Anaphase lagging mainly explains chromosomal mosaicism in human preimplantation embryos. Hum. Reprod. 2004, 19, 316–324. [Google Scholar] [CrossRef]
- Bielanska, M.; Jin, S.; Bernier, M.; Tan, S.L.; Ao, A. Diploid-aneuploid mosaicism in human embryos cultured to the blastocyst stage. Fertil. Steril. 2005, 84, 336–342. [Google Scholar] [CrossRef]
- Taylor, T.H.; Gitlin, S.A.; Patrick, J.L.; Crain, J.L.; Wilson, J.M.; Griffin, D.K. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 2014, 20, 571–581. [Google Scholar] [CrossRef]
- Munne, S.; Blazek, J.; Large, M.; Martinez-Ortiz, P.A.; Nisson, H.; Liu, E.; Tarozzi, N.; Borini, A.; Becker, A.; Zhang, J.; et al. Detailed investigation into the cytogenetic constitution and pregnancy outcome of replacing mosaic blastocysts detected with the use of high-resolution next-generation sequencing. Fertil. Steril. 2017, 108, 62–71.e8. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hao, Y.; Elshewy, N.; Zhu, X.; Zhang, Z.; Zhou, P. The mechanisms and clinical application of mosaicism in preimplantation embryos. J. Assist. Reprod. Genet. 2020, 37, 497–508. [Google Scholar] [CrossRef]
- Chavez, S.L.; Loewke, K.E.; Han, J.; Moussavi, F.; Colls, P.; Munne, S.; Behr, B.; Reijo Pera, R.A. Dynamic blastomere behaviour reflects human embryo ploidy by the four-cell stage. Nat. Commun. 2012, 3, 1251. [Google Scholar] [CrossRef] [Green Version]
- Hassold, T.; Hall, H.; Hunt, P. The origin of human aneuploidy: Where we have been, where we are going. Hum. Mol. Genet. 2007, 16, R203–R208. [Google Scholar] [CrossRef]
- Nikitina, T.V.; Sazhenova, E.A.; Zhigalina, D.I.; Tolmacheva, E.N.; Sukhanova, N.N.; Lebedev, I.N. Karyotype evaluation of repeated abortions in primary and secondary recurrent pregnancy loss. J. Assist. Reprod. Genet. 2020, 37, 517–525. [Google Scholar] [CrossRef]
- Lebedev, I. Mosaic aneuploidy in early fetal losses. Cytogenet. Genome Res. 2011, 133, 169–183. [Google Scholar] [CrossRef]
- Lund, I.C.B.; Becher, N.; Christensen, R.; Petersen, O.B.; Steffensen, E.H.; Vestergaard, E.M.; Vogel, I. Prevalence of mosaicism in uncultured chorionic villus samples after chromosomal microarray and clinical outcome in pregnancies affected by confined placental mosaicism. Prenat. Diagn. 2020, 40, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, T.; Singh, A.K.; Chen, T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 2015, 7, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Tajima, S.; Suetake, I.; Takeshita, K.; Nakagawa, A.; Kimura, H. Domain structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA methyltransferases. Adv. Exp. Med. Biol. 2016, 945, 63–86. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Pradhan, S.; Bacolla, A.; Wells, R.D.; Roberts, R.J. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef] [Green Version]
- Jeanblanc, M.; Mousli, M.; Hopfner, R.; Bathami, K.; Martinet, N.; Abbady, A.Q.; Siffert, J.C.; Mathieu, E.; Muller, C.D.; Bronner, C. The retinoblastoma gene and its product are targeted by ICBP90: A key mechanism in the G1/S transition during the cell cycle. Oncogene 2005, 24, 7337–7345. [Google Scholar] [CrossRef] [Green Version]
- Muto, M.; Kanari, Y.; Kubo, E.; Takabe, T.; Kurihara, T.; Fujimori, A.; Tatsumi, K. Targeted disruption of Np95 gene renders murine embryonic stem cells hypersensitive to DNA damaging agents and DNA replication blocks. J. Biol. Chem. 2002, 277, 34549–34555. [Google Scholar] [CrossRef] [Green Version]
- Papait, R.; Pistore, C.; Negri, D.; Pecoraro, D.; Cantarini, L.; Bonapace, I.M. Np95 is implicated in pericentromeric heterochromatin replication and in major satellite silencing. Mol. Biol. Cell 2007, 18, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meilinger, D.; Fellinger, K.; Bultmann, S.; Rothbauer, U.; Bonapace, I.M.; Klinkert, W.E.; Spada, F.; Leonhardt, H. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep. 2009, 10, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012, 8, 328–330. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Liu, Y.; Upadhyay, A.K.; Chang, Y.; Howerton, S.B.; Vertino, P.M.; Zhang, X.; Cheng, X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012, 40, 4841–4849. [Google Scholar] [CrossRef]
- Oakberg, E.F. Duration of spermatogenesis in the mouse. Nature 1957, 180, 1137–1138. [Google Scholar] [CrossRef]
- Lane, S.; Kauppi, L. Meiotic spindle assembly checkpoint and aneuploidy in males versus females. Cell. Mol. Life Sci. CMLS 2019, 76, 1135–1150. [Google Scholar] [CrossRef] [Green Version]
- Kagiwada, S.; Kurimoto, K.; Hirota, T.; Yamaji, M.; Saitou, M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013, 32, 340–353. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Matsui, Y. Epigenetic events in mammalian germ-cell development: Reprogramming and beyond. Nat. Rev. Genet. 2008, 9, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Yamaji, M. Primordial germ cells in mice. Cold Spring Harb. Perspect. Biol. 2012, 4, a008375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, S.K.; Feil, R. Epigenetic transitions in germ cell development and meiosis. Dev. Cell 2010, 19, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, T.L.; Yang, G.J.; McCarrey, J.R.; Bartolomei, M.S. The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development. Hum. Mol. Genet. 2000, 9, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Henckel, A.; Nakabayashi, K.; Sanz, L.A.; Feil, R.; Hata, K.; Arnaud, P. Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals. Hum. Mol. Genet. 2009, 18, 3375–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Yan, L.; Guo, H.; Li, L.; Hu, B.; Zhao, Y.; Yong, J.; Hu, Y.; Wang, X.; Wei, Y.; et al. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell 2015, 161, 1437–1452. [Google Scholar] [CrossRef] [Green Version]
- Marquez, C.; Egozcue, J.; Martorell, M.R.; Moreno, V.; Templado, C. Colcemid increases the frequency of chromosome abnormalities in human sperm. Cytogenet. Genome Res. 1996, 72, 164–170. [Google Scholar] [CrossRef]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Scelfo, A.; Fachinetti, D. Keeping the centromere under control: A promising role for DNA methylation. Cells 2019, 8, 912. [Google Scholar] [CrossRef] [Green Version]
- Sears, D.D.; Hegemann, J.H.; Shero, J.H.; Hieter, P. Cis-acting determinants affecting centromere function, sister-chromatid cohesion and reciprocal recombination during meiosis in Saccharomyces cerevisiae. Genetics 1995, 139, 1159–1173. [Google Scholar] [PubMed]
- Yelina, N.E.; Choi, K.; Chelysheva, L.; Macaulay, M.; de Snoo, B.; Wijnker, E.; Miller, N.; Drouaud, J.; Grelon, M.; Copenhaver, G.P.; et al. Epigenetic remodeling of meiotic crossover frequency in Arabidopsis thaliana DNA methyltransferase mutants. PLoS Genet. 2012, 8, e1002844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, F.E.; Hornick, J.E.; Lampson, M.A.; Schultz, R.M.; Shea, L.D.; Woodruff, T.K. Chromosome cohesion decreases in human eggs with advanced maternal age. Aging Cell 2012, 11, 1121–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinska, A.P.; Holubcova, Z.; Blayney, M.; Elder, K.; Schuh, M. Sister kinetochore splitting and precocious disintegration of bivalents could explain the maternal age effect. eLife 2015, 4, e11389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.; Tan, S.L.; Hartshorne, G.M.; McAinsh, A.D. Unique geometry of sister kinetochores in human oocytes during meiosis I may explain maternal age-associated increases in chromosomal abnormalities. Biol. Open 2015, 5, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Lagirand-Cantaloube, J.; Ciabrini, C.; Charrasse, S.; Ferrieres, A.; Castro, A.; Anahory, T.; Lorca, T. Loss of centromere cohesion in aneuploid human oocytes correlates with decreased kinetochore localization of the sac proteins Bub1 and Bubr1. Sci. Rep. 2017, 7, 44001. [Google Scholar] [CrossRef]
- Hassold, T.; Hansen, T.; Hunt, P.; VandeVoort, C. Cytological studies of recombination in rhesus males. Cytogenet. Genome Res. 2009, 124, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Ben Khelifa, M.; Ghieh, F.; Boudjenah, R.; Hue, C.; Fauvert, D.; Dard, R.; Garchon, H.J.; Vialard, F. A MEI1 homozygous missense mutation associated with meiotic arrest in a consanguineous family. Hum. Reprod. 2018, 33, 1034–1037. [Google Scholar] [CrossRef]
- Li, B.; Wu, W.; Luo, H.; Liu, Z.; Liu, H.; Li, Q.; Pan, Z. Molecular characterization and epigenetic regulation of Mei1 in cattle and cattle-yak. Gene 2015, 573, 50–56. [Google Scholar] [CrossRef]
- Kuznetsov, S.; Pellegrini, M.; Shuda, K.; Fernandez-Capetillo, O.; Liu, Y.; Martin, B.K.; Burkett, S.; Southon, E.; Pati, D.; Tessarollo, L.; et al. RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J. Biol. 2007, 176, 581–592. [Google Scholar] [CrossRef]
- Hansmann, T.; Pliushch, G.; Leubner, M.; Kroll, P.; Endt, D.; Gehrig, A.; Preisler-Adams, S.; Wieacker, P.; Haaf, T. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum. Mol. Genet. 2012, 21, 4669–4679. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Guo, H.; Ren, Y.; Hou, Y.; Dong, J.; Li, R.; Lian, Y.; Fan, X.; Hu, B.; Gao, Y.; et al. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat. Genet. 2018, 50, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Zhang, Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 2011, 334, 194. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Shen, L.; Dai, Q.; He, C.; Zhang, Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011, 21, 1670–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, C.Y.; Bestor, T.H.; Ding, F.; Latham, K.E.; Mertineit, C.; Trasler, J.M.; Chaillet, J.R. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 2001, 104, 829–838. [Google Scholar] [CrossRef]
- Hirasawa, R.; Chiba, H.; Kaneda, M.; Tajima, S.; Li, E.; Jaenisch, R.; Sasaki, H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008, 22, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Yu, Y.; Fan, Y.; Li, C.; Xu, X.; Duan, J.; Li, R.; Kang, X.; Ma, X.; Chen, X.; et al. Genome wide abnormal DNA methylome of human blastocyst in assisted reproductive technology. J. Genet. Genom. 2017, 44, 475–481. [Google Scholar] [CrossRef]
- McCallie, B.R.; Parks, J.C.; Patton, A.L.; Griffin, D.K.; Schoolcraft, W.B.; Katz-Jaffe, M.G. Hypomethylation and Genetic Instability in Monosomy Blastocysts May Contribute to Decreased Implantation Potential. PLoS ONE 2016, 11, e0159507. [Google Scholar] [CrossRef]
- Braude, P.; Bolton, V.; Moore, S. Human gene expression first occurs between the four- and eight-cell stages of preimplantation development. Nature 1988, 332, 459–461. [Google Scholar] [CrossRef]
- Cao, Y.; Li, M.; Liu, F.; Ni, X.; Wang, S.; Zhang, H.; Sui, X.; Huo, R. Deletion of maternal UHRF1 severely reduces mouse oocyte quality and causes developmental defects in preimplantation embryos. FASEB J. 2019, 33, 8294–8305. [Google Scholar] [CrossRef] [PubMed]
- Samans, B.; Yang, Y.; Krebs, S.; Sarode, G.V.; Blum, H.; Reichenbach, M.; Wolf, E.; Steger, K.; Dansranjavin, T.; Schagdarsurengin, U. Uniformity of nucleosome preservation pattern in Mammalian sperm and its connection to repetitive DNA elements. Dev. Cell 2014, 30, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Colaco, S.; Sakkas, D. Paternal factors contributing to embryo quality. J. Assist. Reprod. Genet. 2018, 35, 1953–1968. [Google Scholar] [CrossRef]
- Sakkas, D.; Urner, F.; Bianchi, P.G.; Bizzaro, D.; Wagner, I.; Jaquenoud, N.; Manicardi, G.; Campana, A. Sperm chromatin anomalies can influence decondensation after intracytoplasmic sperm injection. Hum. Reprod. 1996, 11, 837–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammoud, S.S.; Purwar, J.; Pflueger, C.; Cairns, B.R.; Carrell, D.T. Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil. Steril. 2010, 94, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Benchaib, M.; Braun, V.; Ressnikof, D.; Lornage, J.; Durand, P.; Niveleau, A.; Guerin, J.F. Influence of global sperm DNA methylation on IVF results. Hum. Reprod. 2005, 20, 768–773. [Google Scholar] [CrossRef] [Green Version]
- Aston, K.I.; Uren, P.J.; Jenkins, T.G.; Horsager, A.; Cairns, B.R.; Smith, A.D.; Carrell, D.T. Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil. Steril. 2015, 104, 1388–1397.e5. [Google Scholar] [CrossRef]
- Morel, F.; Roux, C.; Bresson, J.L. Disomy frequency estimated by multicolour fluorescence in situ hybridization, degree of nuclear maturity and teratozoospermia in human spermatozoa. Reproduction 2001, 121, 783–789. [Google Scholar] [CrossRef]
- Ovari, L.; Sati, L.; Stronk, J.; Borsos, A.; Ward, D.C.; Huszar, G. Double probing individual human spermatozoa: Aniline blue staining for persistent histones and fluorescence in situ hybridization for aneuploidies. Fertil. Steril. 2010, 93, 2255–2261. [Google Scholar] [CrossRef]
- Benchaib, M.; Braun, V.; Lornage, J.; Hadj, S.; Salle, B.; Lejeune, H.; Guerin, J.F. Sperm DNA fragmentation decreases the pregnancy rate in an assisted reproductive technique. Hum. Reprod. 2003, 18, 1023–1028. [Google Scholar] [CrossRef]
- Vozdova, M.; Heracek, J.; Sobotka, V.; Rubes, J. Testicular sperm aneuploidy in non-obstructive azoospermic patients. Hum. Reprod. 2012, 27, 2233–2239. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ferreyra, J.; Luna, D.; Villegas, L.; Romero, R.; Zavala, P.; Hilario, R.; Duenas-Chacon, J. High aneuploidy rates observed in embryos derived from donated oocytes are related to male aging and high percentages of sperm DNA fragmentation. Clin. Med. Insights Reprod. Health 2015, 9, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ferreyra, J.; Hilario, R.; Duenas, J. High percentages of embryos with 21, 18 or 13 trisomy are related to advanced paternal age in donor egg cycles. JBRA Assist. Reprod. 2018, 22, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Seli, E.; Gardner, D.K.; Schoolcraft, W.B.; Moffatt, O.; Sakkas, D. Extent of nuclear DNA damage in ejaculated spermatozoa impacts on blastocyst development after in vitro fertilization. Fertil. Steril. 2004, 82, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.G.; Aston, K.I.; Pflueger, C.; Cairns, B.R.; Carrell, D.T. Age-associated sperm DNA methylation alterations: Possible implications in offspring disease susceptibility. PLoS Genet. 2014, 10, e1004458. [Google Scholar] [CrossRef]
- Takeda, K.; Kobayashi, E.; Nishino, K.; Imai, A.; Adachi, H.; Hoshino, Y.; Iwao, K.; Akagi, S.; Kaneda, M.; Watanabe, S. Age-related changes in DNA methylation levels at CpG sites in bull spermatozoa and in vitro fertilization-derived blastocyst-stage embryos revealed by combined bisulfite restriction analysis. J. Reprod. Dev. 2019, 65, 305–312. [Google Scholar] [CrossRef] [Green Version]
- El-Domyati, M.M.; Al-Din, A.B.; Barakat, M.T.; El-Fakahany, H.M.; Xu, J.; Sakkas, D. Deoxyribonucleic acid repair and apoptosis in testicular germ cells of aging fertile men: The role of the poly(adenosine diphosphate-ribosyl)ation pathway. Fertil. Steril. 2009, 91, 2221–2229. [Google Scholar] [CrossRef]
- Robinson, W.P.; Price, E.M. The human placental methylome. Cold Spring Harb. Perspect. Biol. 2015, 5, a023044. [Google Scholar] [CrossRef]
- Weier, J.F.; Weier, H.U.; Jung, C.J.; Gormley, M.; Zhou, Y.; Chu, L.W.; Genbacev, O.; Wright, A.A.; Fisher, S.J. Human cytotrophoblasts acquire aneuploidies as they differentiate to an invasive phenotype. Dev. Biol. 2005, 279, 420–432. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.H.; Wang, H.T.; Kong, Q.P. Dynamic DNA methylation during aging: A “prophet” of age-related outcomes. Front. Genet. 2019, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.; Bonassi, S. The effect of age, gender, diet and lifestyle on DNA damage measured using micronucleus frequency in human peripheral blood lymphocytes. Mutagenesis 2011, 26, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Potter, H.; Chial, H.J.; Caneus, J.; Elos, M.; Elder, N.; Borysov, S.; Granic, A. Chromosome instability and mosaic aneuploidy in neurodegenerative and neurodevelopmental disorders. Front. Genet. 2019, 10, 1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.H. Organisers & Genes; Cambridge University Press: Cambridge, UK, 1940. [Google Scholar]
- Alvarez-Nava, F.; Lanes, R. Epigenetics in turner syndrome. Clin. Epigenet. 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Jamil, M.A.; Nuesgen, N.; Schreiner, F.; Priebe, L.; Hoffmann, P.; Herns, S.; Nothen, M.M.; Frohlich, H.; Oldenburg, J.; et al. DNA methylation signature in peripheral blood reveals distinct characteristics of human X chromosome numerical aberrations. Clin. Epigenet. 2015, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Trolle, C.; Nielsen, M.M.; Skakkebaek, A.; Lamy, P.; Vang, S.; Hedegaard, J.; Nordentoft, I.; Orntoft, T.F.; Pedersen, J.S.; Gravholt, C.H. Widespread DNA hypomethylation and differential gene expression in Turner syndrome. Sci. Rep. 2016, 6, 34220. [Google Scholar] [CrossRef] [Green Version]
- Cotton, A.M.; Ge, B.; Light, N.; Adoue, V.; Pastinen, T.; Brown, C.J. Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome Biol. 2013, 14, R122. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Lee, Y.K.; Lim, Y.C.; Zheng, Z.; Lin, X.M.; Ng, D.P.; Holbrook, J.D.; Law, H.Y.; Kwek, K.Y.; Yeo, G.S.; et al. Global DNA hypermethylation in down syndrome placenta. PLoS Genet. 2013, 9, e1003515. [Google Scholar] [CrossRef] [Green Version]
- Kerkel, K.; Schupf, N.; Hatta, K.; Pang, D.; Salas, M.; Kratz, A.; Minden, M.; Murty, V.; Zigman, W.B.; Mayeux, R.P.; et al. Altered DNA methylation in leukocytes with trisomy 21. PLoS Genet. 2010, 6, e1001212. [Google Scholar] [CrossRef] [Green Version]
- Hatt, L.; Aagaard, M.M.; Bach, C.; Graakjaer, J.; Sommer, S.; Agerholm, I.E.; Kolvraa, S.; Bojesen, A. Microarray-Based Analysis of Methylation of 1st Trimester Trisomic Placentas from Down Syndrome, Edwards Syndrome and Patau Syndrome. PLoS ONE 2016, 11, e0160319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolmacheva, E.N.; Kashevarova, A.A.; Skryabin, N.A.; Lebedev, I.N. Epigenetic effects of trisomy 16 in human placenta. Mol. Biol. 2013, 47, 373–381. [Google Scholar] [CrossRef]
- Blair, J.D.; Langlois, S.; McFadden, D.E.; Robinson, W.P. Overlapping DNA methylation profile between placentas with trisomy 16 and early-onset preeclampsia. Placenta 2014, 35, 216–222. [Google Scholar] [CrossRef]
- Tolmacheva, E.N.; Kashevarova, A.A.; Sukhanova, N.N.; Sazhenova, E.A.; Lebedev, I.N. Epigenetic inactivation of the RB1 gene as a factor of genomic instability: A possible contribution to etiology of chromosomal mosaicism during human embryo development. Russ. J. Genet. 2008, 44, 1266–1271. [Google Scholar] [CrossRef]
- Kashevarova, A.A.; Tolmacheva, E.N.; Sukhanova, N.N.; Sazhenova, E.A.; Lebedev, I.N. Estimation of the mehylation status of the promoter region of the cell cycle control gene P14ARF in placental tissues of spontaneous abortions with chromosomal mosaicism. Russ. J. Genet. 2009, 45, 749–755. [Google Scholar] [CrossRef]
- Lentini, L.; Pipitone, L.; Di Leonardo, A. Functional inactivation of pRB results in aneuploid mammalian cells after release from a mitotic block. Neoplasia 2002, 4, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Iovino, F.; Lentini, L.; Amato, A.; Di Leonardo, A. RB acute loss induces centrosome amplification and aneuploidy in murine primary fibroblasts. Mol. Cancer 2006, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Barra, V.; Schillaci, T.; Lentini, L.; Costa, G.; Di Leonardo, A. Bypass of cell cycle arrest induced by transient DNMT1 post-transcriptional silencing triggers aneuploidy in human cells. Cell Div. 2012, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpf, A.R.; Matsui, S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Hevi, S.; Gay, F.; Tsujimoto, N.; He, T.; Zhang, B.; Ueda, Y.; Li, E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 2007, 39, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.E.; Okano, M.; Dick, F.; Tsujimoto, N.; Chen, T.; Wang, S.; Ueda, Y.; Dyson, N.; Li, E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005, 280, 17986–17991. [Google Scholar] [CrossRef] [Green Version]
- Schueler, M.G.; Sullivan, B.A. Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genom. Hum. Genet. 2006, 7, 301–313. [Google Scholar] [CrossRef]
- Pironon, N.; Puechberty, J.; Roizes, G. Molecular and evolutionary characteristics of the fraction of human α satellite DNA associated with CENP-A at the centromeres of chromosomes 1, 5, 19, and 21. BMC Genom. 2010, 11, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieni, R.S.; Chan, G.K.; Hendzel, M.J. Epigenetics regulate centromere formation and kinetochore function. J. Cell. Biochem. 2008, 104, 2027–2039. [Google Scholar] [CrossRef]
- Heit, R.; Rattner, J.B.; Chan, G.K.; Hendzel, M.J. G2 histone methylation is required for the proper segregation of chromosomes. J. Cell Sci. 2009, 122, 2957–2968. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Yamazaki, T.; Miki, H.; Ogonuki, N.; Inoue, K.; Ogura, A.; Baba, T. Centromeric DNA hypomethylation as an epigenetic signature discriminates between germ and somatic cell lineages. Dev. Biol. 2007, 312, 419–426. [Google Scholar] [CrossRef]
- Pendina, A.A.; Efimova, O.A.; Fedorova, I.D.; Leont’eva, O.A.; Shilnikova, E.M.; Lezhnina, J.G.; Kuznetzova, T.V.; Baranov, V.S. DNA methylation patterns of metaphase chromosomes in human preimplantation embryos. Cytogenet. Genome Res. 2011, 132, 1–7. [Google Scholar] [CrossRef]
- Guttenbach, M.; Schmid, M. Exclusion of specific human chromosomes into micronuclei by 5-azacytidine treatment of lymphocyte cultures. Exp. Cell Res. 1994, 211, 127–132. [Google Scholar] [CrossRef]
- Prada, D.; Gonzalez, R.; Sanchez, L.; Castro, C.; Fabian, E.; Herrera, L.A. Satellite 2 demethylation induced by 5-azacytidine is associated with missegregation of chromosomes 1 and 16 in human somatic cells. Mutat. Res. 2012, 729, 100–105. [Google Scholar] [CrossRef]
- Costa, G.; Barra, V.; Lentini, L.; Cilluffo, D.; Di Leonardo, A. DNA demethylation caused by 5-Aza-2’-deoxycytidine induces mitotic alterations and aneuploidy. Oncotarget 2016, 7, 3726–3739. [Google Scholar] [CrossRef] [Green Version]
- Ostertag, E.M.; Kazazian, H.H., Jr. Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 2001, 35, 501–538. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Bourc’his, D. Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev. 2008, 22, 970–975. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef]
- Schueler, M.G.; Dunn, J.M.; Bird, C.P.; Ross, M.T.; Viggiano, L.; Rocchi, M.; Willard, H.F.; Green, E.D. Progressive proximal expansion of the primate X chromosome centromere. Proc. Natl. Acad. Sci. USA 2005, 102, 10563–10568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schueler, M.G.; Higgins, A.W.; Rudd, M.K.; Gustashaw, K.; Willard, H.F. Genomic and genetic definition of a functional human centromere. Science 2001, 294, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chueh, A.C.; Northrop, E.L.; Brettingham-Moore, K.H.; Choo, K.H.; Wong, L.H. LINE retrotransposon RNA is an essential structural and functional epigenetic component of a core neocentromeric chromatin. PLoS Genet. 2009, 5, e1000354. [Google Scholar] [CrossRef] [Green Version]
- Chueh, A.C.; Wong, L.H.; Wong, N.; Choo, K.H. Variable and hierarchical size distribution of L1-retroelement-enriched CENP-A clusters within a functional human neocentromere. Hum. Mol. Genet. 2005, 14, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Fritz, B.; Hasson, D.; Abrusan, G.; Cheung, F.; Yoda, K.; Radlwimmer, B.; Ladurner, A.G.; Warburton, P.E. Co-localization of CENP-C and CENP-H to discontinuous domains of CENP-A chromatin at human neocentromeres. Genome Biol. 2007, 8, R148. [Google Scholar] [CrossRef] [Green Version]
- Peaston, A.E.; Evsikov, A.V.; Graber, J.H.; de Vries, W.N.; Holbrook, A.E.; Solter, D.; Knowles, B.B. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell 2004, 7, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012, 487, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malki, S.; van der Heijden, G.W.; O’Donnell, K.A.; Martin, S.L.; Bortvin, A. A role for retrotransposon LINE-1 in fetal oocyte attrition in mice. Dev. Cell 2014, 29, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daughtry, B.L.; Chavez, S.L. Chromosomal instability in mammalian pre-implantation embryos: Potential causes, detection methods, and clinical consequences. Cell Tissue Res. 2016, 363, 201–225. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolmacheva, E.N.; Vasilyev, S.A.; Lebedev, I.N. Aneuploidy and DNA Methylation as Mirrored Features of Early Human Embryo Development. Genes 2020, 11, 1084. https://doi.org/10.3390/genes11091084
Tolmacheva EN, Vasilyev SA, Lebedev IN. Aneuploidy and DNA Methylation as Mirrored Features of Early Human Embryo Development. Genes. 2020; 11(9):1084. https://doi.org/10.3390/genes11091084
Chicago/Turabian StyleTolmacheva, Ekaterina N., Stanislav A. Vasilyev, and Igor N. Lebedev. 2020. "Aneuploidy and DNA Methylation as Mirrored Features of Early Human Embryo Development" Genes 11, no. 9: 1084. https://doi.org/10.3390/genes11091084
APA StyleTolmacheva, E. N., Vasilyev, S. A., & Lebedev, I. N. (2020). Aneuploidy and DNA Methylation as Mirrored Features of Early Human Embryo Development. Genes, 11(9), 1084. https://doi.org/10.3390/genes11091084