Harnessing DNA Replication Stress for Novel Cancer Therapy


Abstract
:1. Introduction
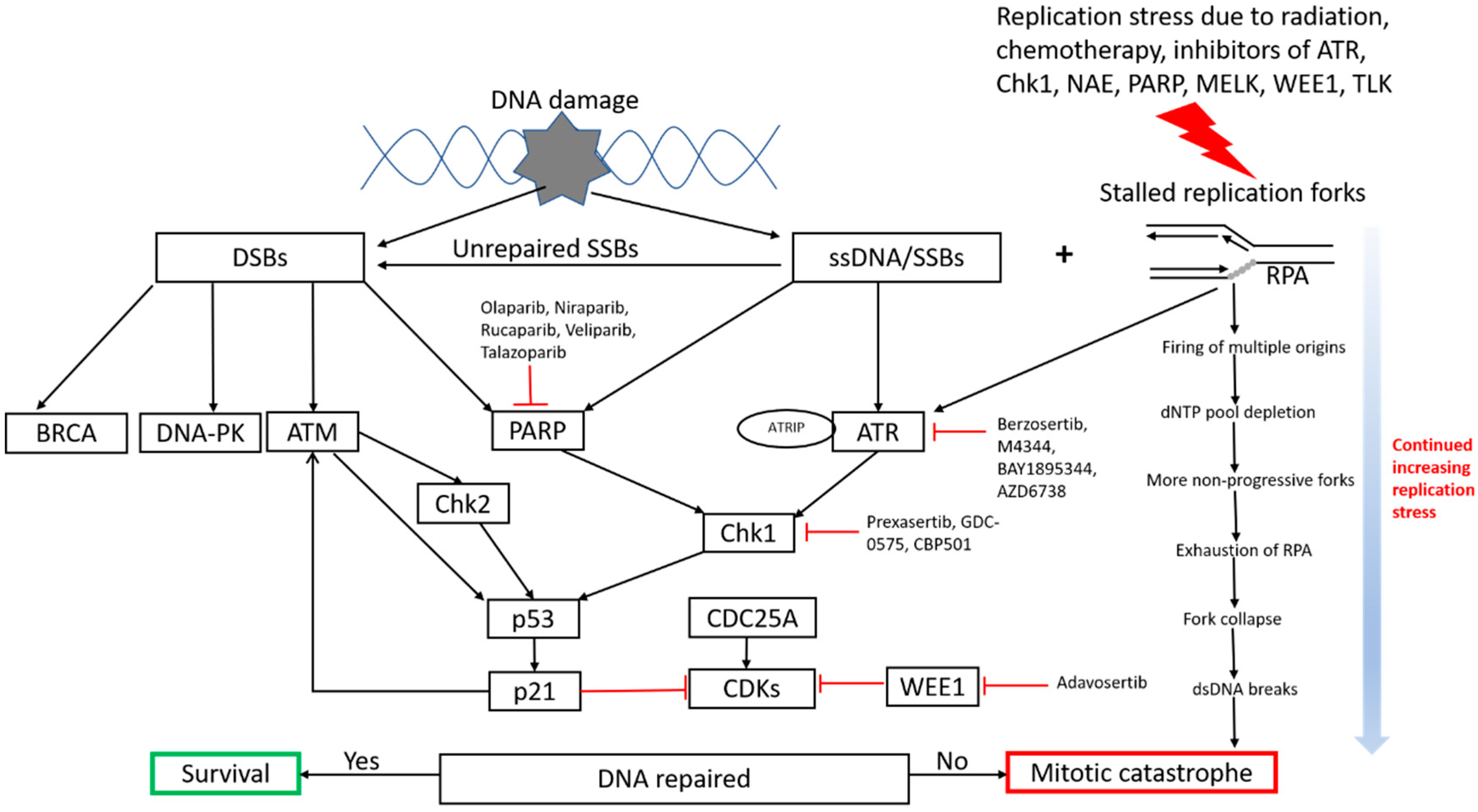
2. Underlying Mechanisms of Replication Stress and Rationale in Cancer Therapy
3. Potential Pathways to Modulate Replication Stress for Cancer Treatment
3.1. Traditional Approaches—Radiation and Chemotherapy
3.2. New Emerging Approaches and Strategies
3.2.1. ATR-Chk1 Pathway
3.2.2. PARP Inhibitors
3.2.3. Other Targets That Are Relevant to DNA Replication Stress
4. Potential Combination Treatment Approaches
5. Future Directions, Identification of Biomarkers and Resistance Mechanisms
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fragkos, M.; Ganier, O.; Coulombe, P.; Mechali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.P.; Patel, C.N.; Sureja, D.K.; Sanghavi, K.P. A Review on DNA Repair Inhibition by PARP Inhibitors in Cancer Therapy. Folia Med. 2018, 60, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Eykelenboom, J.K.; Harte, E.C.; Canavan, L.; Pastor-Peidro, A.; Calvo-Asensio, I.; Llorens-Agost, M. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013, 5, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell 2017, 66, 735–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canman, C.E. Replication checkpoint: Preventing mitotic catastrophe. Curr. Biol. 2001, 11, R121–R124. [Google Scholar] [CrossRef] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Replication stress and cancer: It takes two to tango. Exp. Cell Res. 2014, 329, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011, 11, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Kotsantis, P.; Jones, R.M.; Higgs, M.R.; Petermann, E. Cancer therapy and replication stress: Forks on the road to perdition. Adv. Clin. Chem. 2015, 69, 91–138. [Google Scholar] [PubMed]
- Ewald, B.; Sampath, D.; Plunkett, W. Nucleoside analogs: Molecular mechanisms signaling cell death. Oncogene 2008, 27, 6522–6537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, D.B.; Harkin, P.D.; Johnston, G.P. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Regairaz, M.; Zhang, Y.W.; Fu, H.; Agama, K.K.; Tata, N.; Agrawal, S.; Aladjem, M.I.; Pommier, Y. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 2011, 195, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, J.A.; Conti, C.; Syed, A.; Aladjem, M.I.; Pommier, Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: Single-cell and -DNA fiber analyses. Mol. Cell Biol. 2007, 27, 5806–5818. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Loegering, D.; Arlander, S.J.; Hackbarth, J.; Vroman, B.T.; Roos-Mattjus, P.; Hopkins, K.M.; Lieberman, H.B.; Karnitz, L.M.; Kaufmann, S.H. Rad9 protects cells from topoisomerase poison-induced cell death. J. Biol. Chem. 2004, 279, 18641–18647. [Google Scholar] [CrossRef] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salovska, B.; Janeckova, H.; Fabrik, I.; Karlikova, R.; Cechakova, L.; Ondrej, M.; Link, M.; Friedecky, D.; Tichy, A. Radio-sensitizing effects of VE-821 and beyond: Distinct phosphoproteomic and metabolomic changes after ATR inhibition in irradiated MOLT-4 cells. PLoS ONE 2018, 13, e0199349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef] [Green Version]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montana, M.F.; Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Hocke, S.; Guo, Y.; Job, A.; Orth, M.; Ziesch, A.; Lauber, K.; de Toni, E.N.; Gress, T.M.; Herbst, A.; Goke, B.; et al. A synthetic lethal screen identifies ATR-inhibition as a novel therapeutic approach for POLD1-deficient cancers. Oncotarget 2016, 7, 7080–7095. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.E.; Fleuren, E.D.G.; Frankum, J.; Konde, A.; Williamson, C.T.; Krastev, D.B.; Pemberton, H.N.; Campbell, J.; Gulati, A.; Elliott, R.; et al. ATR Is a Therapeutic Target in Synovial Sarcoma. Cancer Res. 2017, 77, 7014–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United States National Library of Medicine at National Institutes of Health. Available online: https://clinicaltrials.gov (accessed on 26 August 2018).
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauge, S.; Naucke, C.; Hasvold, G.; Joel, M.; Rodland, G.E.; Juzenas, P.; Stokke, T.; Syljuasen, R.G. Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget 2017, 8, 10966–10979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell. 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 2014, 40, 109–117. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar]
- De Klein, A.; Muijtjens, M.; van Os, R.; Verhoeven, Y.; Smit, B.; Carr, A.M.; Lehmann, A.R. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000, 10, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Fang, B. Development of synthetic lethality anticancer therapeutics. J. Med. Chem. 2014, 57, 7859–7873. [Google Scholar] [CrossRef]
- Fuse, E.; Tanii, H.; Kurata, N.; Kobayashi, H.; Shimada, Y.; Tamura, T.; Sasaki, Y.; Tanigawara, Y.; Lush, R.D.; Headlee, D.; et al. Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human alpha1-acid glycoprotein. Cancer Res. 1998, 58, 3248–3253. [Google Scholar]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Ahel, I.; Ahel, D.; Matsusaka, T.; Clark, A.J.; Pines, J.; Boulton, S.J.; West, S.C. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008, 451, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, W.; Bruhn, C.; Grigaravicius, P.; Zhou, Z.W.; Li, F.; Kruger, A.; Siddeek, B.; Greulich, K.O.; Popp, O.; Meisezahl, C.; et al. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 2013, 4, 2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benafif, S.; Hall, M. An update on PARP inhibitors for the treatment of cancer. Onco. Targets Ther. 2015, 8, 519. [Google Scholar]
- Ohmoto, A.; Yachida, S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco. Targets Ther. 2017, 10, 5195–5208. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Liu, C.; Srihari, S.; Cao, K.A.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, P.W.; Jacobson, E.L.; Benjamin, R.C.; Moss, J.; Jacobson, M.K. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J. Biol. Chem. 1989, 264, 4312–4317. [Google Scholar]
- Chen, G.; Zeller, W.J. Multiple effects of 3-aminobenzamide on DNA damage induced by cisplatin (DDP) in DDP-sensitive and -resistant rat ovarian tumor cell lines. Cancer Lett. 1992, 67, 27–33. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Pang, Y.; Pacak, K.; Yang, C. Double-barreled gun: Combination of PARP inhibitor with conventional chemotherapy. Pharmacol. Ther. 2018, 188, 168–175. [Google Scholar] [CrossRef]
- Hengel, S.R.; Malacaria, E.; Constantino, L.F.d.; Bain, F.E.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife 2016, 5, e14740. [Google Scholar] [CrossRef]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [Green Version]
- Manthei, K.A.; Keck, J.L. The BLM dissolvasome in DNA replication and repair. Cell Mol. Life Sci. 2013, 70, 4067–4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Popuri, V.; Croteau, D.L.; Brosh, R.M., Jr.; Bohr, V.A. RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle 2012, 11, 4252–4265. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Brosh, R.M., Jr. RECQL: A new breast cancer susceptibility gene. Cell Cycle 2015, 14, 3540–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathaneni, S.; Sharma, S. The DNA repair helicase RECQ1 has a checkpoint-dependent role in mediating DNA damage responses induced by gemcitabine. J. Biol. Chem. 2019, 294, 15330–15345. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.M.; Galvan, A.M.; Imamura-Kawasawa, Y.; Moldovan, G.L.; Nicolae, C.M. PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucleic Acids Res. 2018, 46, 8908–8916. [Google Scholar] [CrossRef] [Green Version]
- Budke, B.; Lv, W.; Kozikowski, A.P.; Connell, P.P. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? Chem. Med. Chem. 2016, 11, 2468–2473. [Google Scholar] [CrossRef] [Green Version]
- Dungrawala, H.; Bhat, K.P.; le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386.e5. [Google Scholar] [CrossRef]
- Krajewska, M.; Fehrmann, R.S.; Schoonen, P.M.; Labib, S.; de Vries, E.G.; Franke, L.; van Vugt, M.A. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene 2015, 34, 3474–3481. [Google Scholar] [CrossRef]
- Kim, D.; Liu, Y.; Oberly, S.; Freire, R.; Smolka, M.B. ATR-mediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells. Nucleic Acids Res. 2018, 46, 8311–8325. [Google Scholar] [CrossRef] [Green Version]
- Lui, G.Y.L.; Grandori, C.; Kemp, C.J. CDK12: An emerging therapeutic target for cancer. J. Clin. Pathol. 2018, 71, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, D.R.; Jonnalagadda, J.C.; Gatei, M.H.; Sillje, H.H.; Zhou, B.B.; Nigg, E.A.; Khanna, K. Suppression of Tousled-like kinase activity after DNA damage or replication block requires ATM, NBS1 and Chk1. Oncogene 2003, 22, 5927–5937. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Segura-Bayona, S.; Villamor-Paya, M.; Saredi, G.; Todd, M.A.M.; Attolini, C.S.; Chang, T.Y.; Stracker, T.H.; Groth, A. Tousled-like kinases stabilize replication forks and show synthetic lethality with checkpoint and PARP inhibitors. Sci. Adv. 2018, 4, eaat4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2018, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Beke, L.; Kig, C.; Linders, J.T.; Boens, S.; Boeckx, A.; van Heerde, E.; Parade, M.; de Bondt, A.; van den Wyngaert, I.; Bashir, T.; et al. MELK-T1, a small-molecule inhibitor of protein kinase MELK, decreases DNA-damage tolerance in proliferating cancer cells. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, B.B.; Li, J.; Roberts, T.M.; Zhao, J.J. A Conditional Dependency on MELK for the Proliferation of Triple-Negative Breast Cancer Cells. iScience 2018, 9, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Paiva, C.; Godbersen, J.C.; Berger, A.; Brown, J.R.; Danilov, A.V. Targeting neddylation induces DNA damage and checkpoint activation and sensitizes chronic lymphocytic leukemia B cells to alkylating agents. Cell Death Dis. 2015, 6, e1807. [Google Scholar] [CrossRef] [Green Version]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Milhollen, M.A.; Narayanan, U.; Soucy, T.A.; Veiby, P.O.; Smith, P.G.; Amidon, B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011, 71, 3042–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.J.; Milhollen, M.A.; Smith, P.G.; Narayanan, U.; Dutta, A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010, 70, 10310–10320. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.; Ashley, A.K.; Hromas, R.; Nickoloff, J.A. More forks on the road to replication stress recovery. J. Mol. Cell Biol. 2011, 3, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef]
- Sher, D.J. Neoadjuvant Chemoradiotherapy for Stage III Non-Small Cell Lung Cancer. Front Oncol. 2017, 7, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, M.; Budach, W. Definitive chemoradiotherapy. J. Thorac. Dis. 2017, 9, S792–S798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schernberg, A.; del Campo, E.R.; Rousseau, B.; Matzinger, O.; Loi, M.; Maingon, P.; Huguet, F. Adjuvant chemoradiation for gastric carcinoma: State of the art and perspectives. Clin. Transl. Radiat. Oncol. 2018, 10, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.V. Developing combination strategies using PD-1 checkpoint inhibitors to treat cancer. Semin. Immunopathol. 2018, 41, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.; Saxena, A.; Schneider, B.J. Systemic therapy for small cell lung cancer. J. Natl. Compr. Canc. Netw. 2013, 11, 780–787. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.P.; Gilmour, L.; et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Flanagan, C.H.; O’Shea, S.; Lyons, A.; Fogarty, F.M.; McCabe, N.; Kennedy, R.D.; O’Connor, R. IGF-1R inhibition sensitizes breast cancer cells to ATM-related kinase (ATR) inhibitor and cisplatin. Oncotarget 2016, 7, 56826–56841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan, S.V.; Bhadury, J.; Nilsson, L.M.; Green, L.C.; McLure, K.G.; Nilsson, J.A. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 2016, 35, 4689–4697. [Google Scholar] [CrossRef]
- Muralidharan, S.V.; Einarsdottir, B.O.; Bhadury, J.; Lindberg, M.F.; Wu, J.; Campeau, E.; Bagge, R.O.; Stierner, U.; Ny, L.; Nilsson, L.M.; et al. BET bromodomain inhibitors synergize with ATR inhibitors in melanoma in melanoma. Cell Death Dis. 2017, 8, e2982. [Google Scholar] [CrossRef] [Green Version]
- Carrassa, L.; Chila, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- Pfister, S.X.; Markkanen, E.; Jiang, Y.; Sarkar, S.; Woodcock, M.; Orlando, G.; Mavrommati, I.; Pai, C.C.; Zalmas, L.P.; Drobnitzky, N.; et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell 2015, 28, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Aarts, M.; Bajrami, I.; Herrera-Abreu, M.T.; Elliott, R.; Brough, R.; Ashworth, A.; Lord, C.J.; Turner, N.C. Functional Genetic Screen Identifies Increased Sensitivity to WEE1 Inhibition in Cells with Defects in Fanconi Anemia and HR Pathways. Mol. Cancer Ther. 2015, 14, 865–876. [Google Scholar] [CrossRef] [Green Version]
- Lallo, A.; Frese, K.K.; Morrow, C.J.; Sloane, R.; Gulati, S.; Schenk, M.W.; Trapani, F.; Simms, N.; Galvin, M.; Brown, S.; et al. The Combination of the PARP Inhibitor Olaparib and the WEE1 Inhibitor AZD1775 as a New Therapeutic Option for Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5153–5164. [Google Scholar] [CrossRef] [Green Version]
- Perez, R.P.; Lewis, L.D.; Beelen, A.P.; Olszanski, A.J.; Johnston, N.; Rhodes, C.H.; Beaulieu, B.; Ernstoff, M.S.; Eastman, A. Modulation of cell cycle progression in human tumors: A pharmacokinetic and tumor molecular pharmacodynamic study of cisplatin plus the Chk1 inhibitor UCN-01 (NSC 638850). Clin. Cancer Res. 2006, 12, 7079–7085. [Google Scholar] [CrossRef] [Green Version]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.B.; Fornari, C.; Yates, J.W.; de Fernandez, S.B.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR inhibitor AZD6738 synergizes with gemcitabine in vitro and in vivo to induce pancreatic ductal adenocarcinoma regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.B.; Courtin, A.; Boyce, R.J.; Boyle, R.G.; Richards, F.M.; Jodrell, D.I. CHK1 Inhibition Synergizes with Gemcitabine Initially by Destabilizing the DNA Replication Apparatus. Cancer Res. 2015, 75, 3583–3595. [Google Scholar] [CrossRef] [Green Version]
- Rajeshkumar, N.V.; de Oliveira, E.; Ottenhof, N.; Watters, J.; Brooks, D.; Demuth, T.; Shumway, S.D.; Mizuarai, S.; Hirai, H.; Maitra, A.; et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin. Cancer Res. 2011, 17, 2799–2806. [Google Scholar] [CrossRef] [Green Version]
- Garcia, K.; Blank, J.L.; Bouck, D.C.; Liu, X.J.; Sappal, D.S.; Hather, G.; Cosmopoulos, K.; Thomas, M.P.; Kuranda, M.; Pickard, M.D.; et al. Nedd8-activating enzyme inhibitor MLN4924 provides synergy with mitomycin C through interactions with ATR, BRCA1/BRCA2, and chromatin dynamics pathways. Mol. Cancer Ther. 2014, 13, 1625–1635. [Google Scholar] [CrossRef] [Green Version]
- Seto, T.; Esaki, T.; Hirai, F.; Arita, S.; Nosaki, K.; Makiyama, A.; Kometani, T.; Fujimoto, C.; Hamatake, M.; Takeoka, H.; et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2013, 72, 619–627. [Google Scholar] [CrossRef]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Keytruda (Pembrolizumab) [Package Insert]. Merck Sharp & Dohme Corp., Whitehouse Station, NJ 08889, USA. Available online: https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf (accessed on 29 July 2018).
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA damage and innate immunity: Links and trade-offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Wells, C.E.; Bhaskara, S.; Stengel, K.R.; Zhao, Y.; Sirbu, B.; Chagot, B.; Cortez, D.; Khabele, D.; Chazin, W.J.; Cooper, A.; et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS ONE 2013, 8, e68915. [Google Scholar] [CrossRef]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [Green Version]
- Conti, C.; Leo, E.; Eichler, G.S.; Sordet, O.; Martin, M.M.; Fan, A.; Aladjem, M.I.; Pommier, Y. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010, 70, 4470–4480. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Patel, A.A.; Tang, L.; Silver, N.L.; Lindemann, A.; Takahashi, H.; Jaksik, R.; Rao, X.; Kalu, N.N.; Chen, T.C.; et al. Replication Stress Leading to Apoptosis within the S-phase Contributes to Synergism between Vorinostat and AZD1775 in HNSCC Harboring High-Risk TP53 Mutation. Clin. Cancer Res. 2017, 23, 6541–6554. [Google Scholar] [CrossRef] [Green Version]
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012, 3, e285. [Google Scholar] [CrossRef] [Green Version]
- Dotiwala, F.; Eapen, V.V.; Harrison, J.C.; Arbel-Eden, A.; Ranade, V.; Yoshida, S.; Haber, J.E. DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc. Natl. Acad. Sci. USA 2013, 110, E41–E49. [Google Scholar] [CrossRef] [Green Version]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Komarova, E.A. Prospective therapeutic applications of p53 inhibitors. Biochem. Biophys. Res. Commun. 2005, 331, 726–736. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Strom, C.E.; Svensson, L.M.; Schultz, N.; Lundback, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Gokturk, C.; Sanjiv, K.; Stromberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, K.M.; Jones, R.M.; Petermann, E.; Jeggo, P.A. Diminished origin-licensing capacity specifically sensitizes tumor cells to replication stress. Mol. Cancer Res. 2013, 11, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Steckel, M.; Molina-Arcas, M.; Weigelt, B.; Marani, M.; Warne, P.H.; Kuznetsov, H.; Kelly, G.; Saunders, B.; Howell, M.; Downward, J.; et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012, 22, 1227–1245. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Chen, L.; Wu, W.; Garribba, L.; Tian, H.; Liu, Z.; Vogel, I.; Li, C.; Hickson, I.D.; Liu, Y. Potential biomarkers of DNA replication stress in cancer. Oncotarget 2017, 8, 36996–37008. [Google Scholar] [CrossRef]
- Gadaleta, M.C.; Iwasaki, O.; Noguchi, C.; Noma, K.; Noguchi, E. Chromatin immunoprecipitation to detect DNA replication and repair factors. Methods Mol. Biol. 2015, 1300, 169–186. [Google Scholar]
- Syljuasen, R.G.; Sorensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Haynes, B.; Murai, J.; Lee, J.M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Oplustil O’Connor, L.; Rulten, S.L.; Cranston, A.N.; Odedra, R.; Brown, H.; Jaspers, J.E.; Jones, L.; Knights, C.; Evers, B.; Ting, A.; et al. The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models. Cancer Res. 2016, 76, 6084–6094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oplustilova, L.; Wolanin, K.; Mistrik, M.; Korinkova, G.; Simkova, D.; Bouchal, J.; Lenobel, R.; Bartkova, J.; Lau, A.; O’Connor, M.J.; et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle 2012, 11, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Restelli, V.; Chila, R.; Lupi, M.; Rinaldi, A.; Kwee, I.; Bertoni, F.; Damia, G.; Carrassa, L. Characterization of a mantle cell lymphoma cell line resistant to the Chk1 inhibitor PF-00477736. Oncotarget 2015, 6, 37229–37240. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Mechanism | Agents | Approved Indications |
|---|---|---|
| DNA mis incorporation/modification | Cyclophosphamide | HL, NHL, multiple myeloma, ALL, AML, breast cancer, CLL, CML, CLL, CML, mycosis fungoides, neuroblastoma, ovarian cancer, retinoblastoma |
| Temozolomide | Anaplastic astrocytoma, glioblastoma multiforme | |
| Cisplatin | Bladder, testicular, ovarian cancer | |
| Carboplatin | Ovarian cancer | |
| Oxaliplatin | Colorectal cancer | |
| Ribonucleotide reductase inhibitor | Gemcitabine | NSCLC, breast, ovarian, pancreatic cancer |
| Clofarabine | ALL | |
| Fludarabine | CLL | |
| Cytarabine | ALL, AML, CML, meningeal leukemia, lymphomatous meningitis | |
| Hydroxyurea | CML, HNSCC | |
| Thymidylate synthetase inhibition | 5-Fluorouracil | Breast, colorectal, gastric, pancreatic cancer |
| Capecitabine | Breast, colorectal caner | |
| Pemetrexed | NSCLC, malignant pleural mesothelioma | |
| Topoisomerase I inhibitor | Irinotecan | Colorectal cancer, pancreatic cancer |
| Topotecan | Cervical, ovarian, SCLC | |
| Topoisomerase II inhibitor | Etoposide | SCLC, testicular cancer |
| Doxorubicin | ALL, AML, HL, NHL, neuroblastoma, SCLC, soft tissue and bone sarcomas, Wilms tumor, thyroid, ovarian, breast, gastric, transitional cell bladder cancer | |
| DNA methyltransferase inhibitor | Decitabine | MDS |
| Folic acid reductase inhibitor | Methotrexate | ALL, gestational trophoblastic disease, mycosis fungoides, NHL, osteosarcoma, head and neck, breast, lung cancer |
| DNA replication inhibitor | Nelarabine | T-cell ALL, T-cell lymphoblastic lymphoma |
| Blockage of synthesis and metabolism of purine nucleotides | Thioguanine | AML |
| Inhibition of nucleotide synthesis and nucleotide analogue incorporation | Trifluridine and Tipiracil Hydrochloride | Colorectal cancer |
| PARP inhibitors | Olaparib | BRCA-mutated, HER2-negative metastatic breast cancer, BRCA mutated advanced ovarian cancer, as maintenance treatment for recurrent epithelial OPFTC in patients experiencing a complete or partial response to platinum-based chemotherapy |
| Rucaparib | BRCA mutated epithelial OPFTC after two or more chemotherapies, maintenance treatment of recurrent epithelial OPFTC that are in a complete or partial response to platinum-based chemotherapy | |
| Niraparib | Maintenance treatment of recurrent epithelial OPFTC in patients undergoing complete or partial response to platinum-based chemotherapy | |
| Talazoparib | Treatment of adult patients with deleterious or suspected deleterious germline breast cancer susceptibility gene (BRCA)-mutated (gBRCAm) human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer |
| Mechanism. | Drug | Phase | Details (Including NCT Number) |
|---|---|---|---|
| ATR inhibitor | M-6620/Berzosertib/VE-822/VX-970 | I | With cisplatin, and radiation in HPV negative HNSCC(NCT02567422), with standard treatment in esophageal and other cancer (NCT03641547), monotherapy or with carboplatin and paclitaxel in advanced solid tumors (NCT03309150), advanced solid tumors (NCT02157792), with irinotecan in advanced solid tumors (NCT02595931), with WBRT in brain metastasis due to NSCLC, SCLC, or NET (NCT02589522) |
| I/II | Carboplatin and gemcitabine in advanced OPPFTC (NCT02627443) | ||
| II | With irinotecan in advanced TP53 mutant gastric or GEJ cancer (NCT03641313), selected tumors (NCT03718091), gemcitabine in recurrent OPPFTC (NCT02595892), cisplatin and gemcitabine in urothelial cancer (NCT02567409), avelumab and carboplatin in PARPi-resistant ovarian cancer (NCT03704467), carboplatin +/-docetaxel in mCRPC (NCT03517969) | ||
| AZD6738 | I | HNSCC (NCT03022409), with paclitaxel in refractory cancers (NCT02630199), alone or with radiation (NCT02223923), with AZD9150 or acalabrutinib in refractory NHL (NCT03527147), with gemcitabine in advanced solid tumors (NCT03669601) | |
| I/II | With carboplatin or olaparib or MEDI4736 in advanced solid malignancies (NCT02264678), with acalabrutinib in CLL (NCT03328273) | ||
| II | In combination with olaparib in SCLC (NCT03428607, NCT02937818), with olaparib in recurrent ovarian cancer (NCT03462342), with olaparib in metastatic triple negative breast cancer (NCT03330847), with olaparib in tumors with mutations in HDR genes (NCT02576444), with durvalumab in NSCLC (NCT03334617), with olaparib in selected tumors (NCT03682289), neoadjuvant chemotherapy resistant TNBC (NCT03740893) | ||
| BAY1895344 | I | Advanced solid tumors and lymphomas (NCT03188965) | |
| VX-803/M4344 | I | Single agent or in combination with cisplatin, carboplatin or gemcitabine in advanced solid tumors (NCT02278250) | |
| Chk1 inhibitor | LY2606368 (Prexasertib) | I | With cytarabine and fludarabine in AML and high risk MDS (NCT02649764), advanced cancer (NCT02778126, NCT02514603, NCT01115790), refractory solid tumors in pediatric patients (NCT02808650), with ralimetinib in selected cancers (NCT02860780), with cisplatin/cetuximab and radiation in HNSCC (NCT02555644), with olaparib in advanced solid tumors (NCT03057145), with LY3300054 in advanced solid tumors (NCT03495323), with chemotherapy or targeted agents in advanced cancer (NCT02124148), with mitoxantrone, etoposide, and cytarabine in refractory AML and high risk MDS (NCT03735446) |
| II | Extensive stage SCLC (NCT02735980), in BRCA1/2 mutated selected cancers (NCT02203513), in solid tumors with replicative stress or HDR deficiency (NCT02873975), refractory ovarian cancer (NCT03414047) | ||
| CBP501 | I | With cisplatin and nivolumab in advanced solid tumors (NCT03113188) | |
| WEE1 | Adavosertib/AZD1775/MK-1775 | I | Advanced solid tumors (NCT01748825, NCT02610075, NCT02482311, NCT03313557), recurrent GBM (NCT02207010), with radiation and temozolomide in GBM (NCT01849146), with olaparib in refractory solid tumors (NCT02511795), with docetaxel and cisplatin before surgery in NSCLC (NCT02508246), with cisplatin and radiation in HNSCC (NCT03028766), radiation and cisplatin in cervical, vaginal or uterine cancer (NCT03345784), pharmacokinetic studies in solid tumors (NCT03333824), with radiation in pontine gliomas in pediatric patients (NCT01922076), with MEDI4736 in solid tumors (NCT02617277), with irinotecan in RAS or BRAF mutated colorectal cancer (NCT02906059), ovarian cancer (NCT02659241), with MEDI4736 in bladder cancer (NCT02546661) |
| I/II | With gemcitabine (+Radiation) in pancreatic adenocarcinoma (NCT02037230), with carboplatin in refractory tumors (NCT02813135), with nab-paclitaxel and gemcitabine in pancreatic cancer (NCT02194829), with irinotecan in refractory solid tumors in younger patients (NCT02095132) | ||
| II | Uterine serous carcinoma (NCT03668340), SCLC (NCT02593019), in solid tumors with CCNE1 amplification (NCT03253679), BRCA mutated tumors (NCT02465060), with carboplatin and paclitaxel in squamous cell lung cancer (NCT02513563), with concurrent radiation and cisplatin in HNSCC (NCT02585973), with gemcitabine in OPFTC (NCT02101775), with cisplatin in breast cancer (NCT03012477), in AML, MDS and myelofibrosis (NCT03718143), with chemotherapy in OPFTC (NCT02272790), with olaparib in metastatic triple negative breast cancer (NCT03330847), SETD2-deficient advanced tumors (NCT03284385), with paclitaxel in advanced TP53 mutated gastric cancer (NCT02448329), prostate cancer (NCT03385655), with or without olaparib in recurrent OPFTC (NCT03579316), with olaparib in advanced solid tumors (NCT02576444), with carboplatin in advanced solid tumors (NCT01827384), with carboplatin in extensive SCLC (NCT02937818) | ||
| MELK | OTS167 | I | Refractory advanced breast cancer (NCT02926690) |
| I/II | Refractory AML, ALL, advanced MDS, MPN, CML (NCT02795520) | ||
| NEDD8 activating enzyme inhibitor | Pevonedistat/TAK-924/MLN4924 | I | Advanced solid tumors (NCT03330106, NCT03486314), with low dose cytarabine in AML and MDS (NCT03459859), with irinotecan and temozolomide in selected tumors (NCT03323034), with ruxolitinib in myelofibrosis (NCT03386214), with decitabine in high risk AML (NCT03009240), with chemotherapy for refractory ALL (NCT03349281), as single agent or with azacytidine in AML and MDS (NCT02782468) |
| I/II | Alone or with chemotherapy in mesothelioma (NCT03319537), with azacytidine in AML (NCT03013998), with cytarabine, and idarubicin in AML (NCT03330821) | ||
| II | With azacytidine in refractory AML (NCT03745352), with azacytidine in high risk MDS, CMML or low blast AML (NCT02610777), with azacytidine as maintenance therapy after allogeneic stem cell transplantation for non-remission AML (NCT03709576), with azacytidine in MDS or MDS/MPN after failure of DNA methyl transferase inhibitors (NCT03238248), with docetaxel in NSCLC (NCT03228186), with ibrutinib in refractory CLL and NHL (NCT03479268) | ||
| III | With azacytidine in high risk MDS, CMML or low blast AML (NCT03268954) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Swami, U.; Preet, R.; Zhang, J. Harnessing DNA Replication Stress for Novel Cancer Therapy. Genes 2020, 11, 990. https://doi.org/10.3390/genes11090990
Zhu H, Swami U, Preet R, Zhang J. Harnessing DNA Replication Stress for Novel Cancer Therapy. Genes. 2020; 11(9):990. https://doi.org/10.3390/genes11090990
Chicago/Turabian StyleZhu, Huanbo, Umang Swami, Ranjan Preet, and Jun Zhang. 2020. "Harnessing DNA Replication Stress for Novel Cancer Therapy" Genes 11, no. 9: 990. https://doi.org/10.3390/genes11090990