Abundance, Diversity and Role of ICEs and IMEs in the Adaptation of Streptococcus salivarius to the Environment

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. S. salivarius Genomes and Phylogenetic Analysis
2.1.1. S. salivarius Strains and Genome Analysis
2.1.2. Phylogenetic Tree Based on Single Nucleotide Polymorphisms (SNPs)
2.2. Detection and Delineation of the Integrative Elements
2.3. Characterization of Cargo Genes Encoded by ICEs and IMEs
3. Results
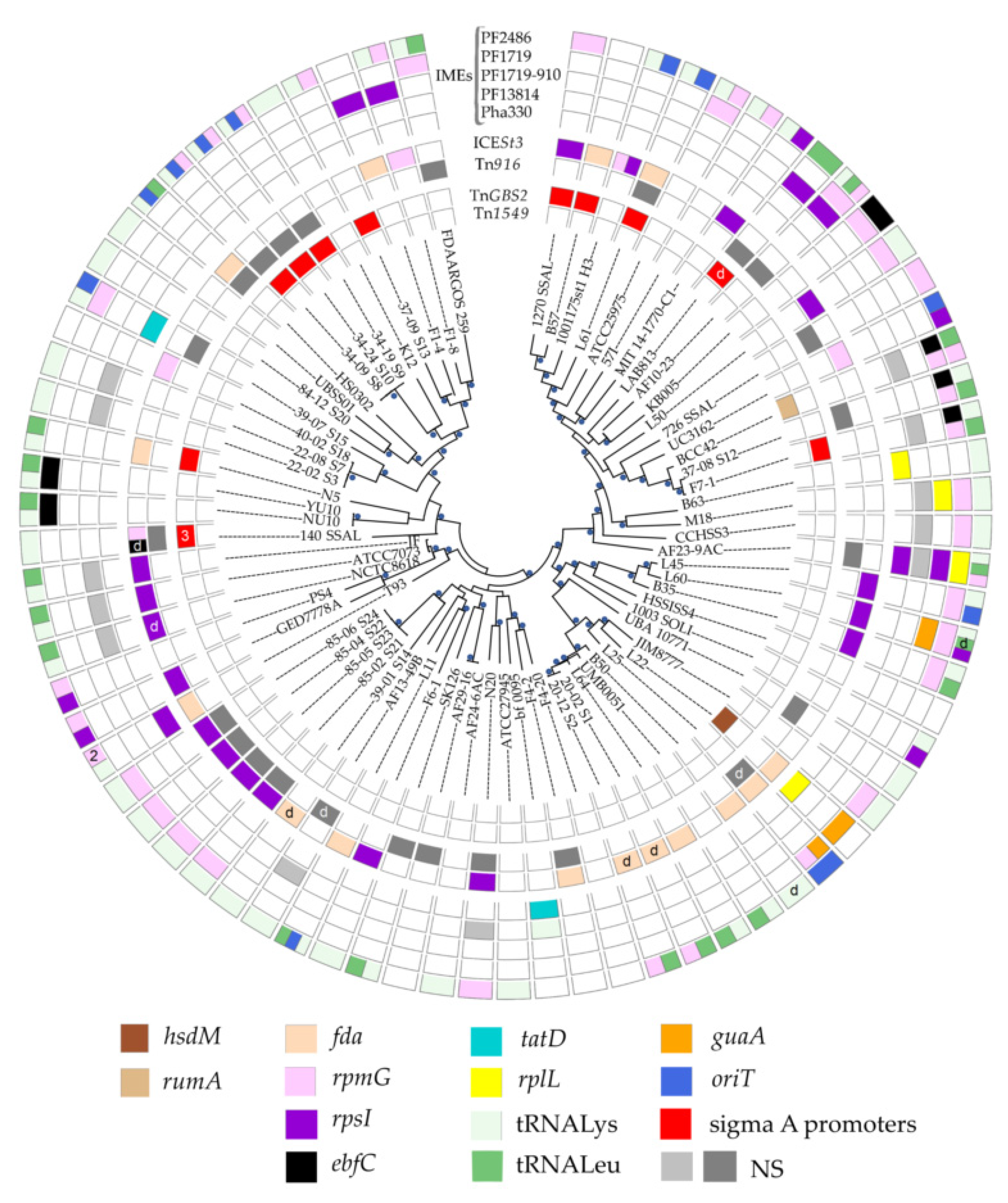
3.1. ICEs in S. salivarius Genomes
3.1.1. ICE Prevalence and Diversity
3.1.2. ICE Integrases and Integration Sites
3.1.3. Slightly Decayed Elements Deriving from ICEs
3.2. IMEs in S. salivarius Genomes
3.2.1. IME Prevalence and Diversity
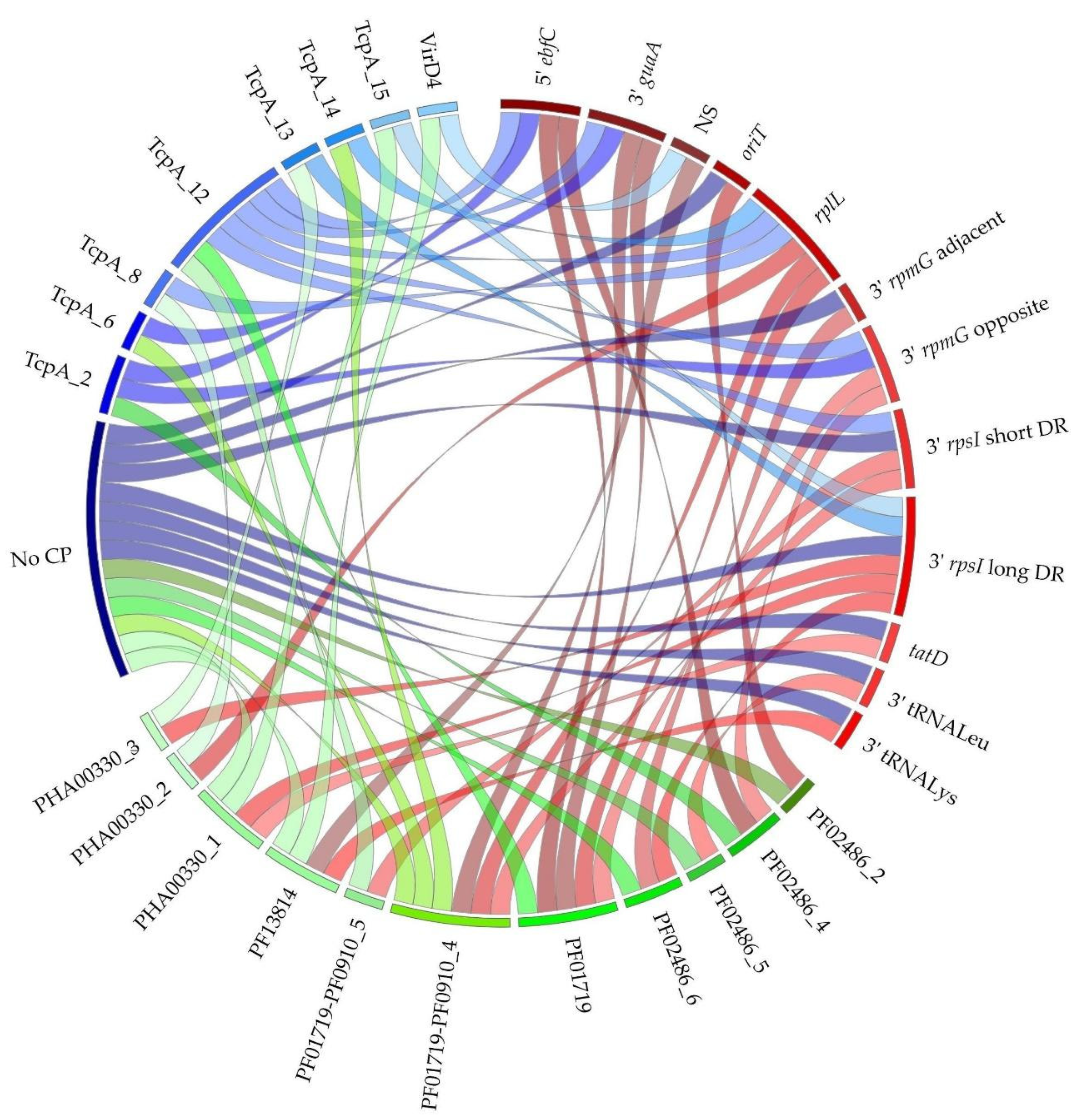
3.2.2. IME Integrases and Integration Sites
3.2.3. Diversity of Integrase-Relaxase-CP Combinations within IMEs
3.3. Composite Elements in S. salivarius Genomes
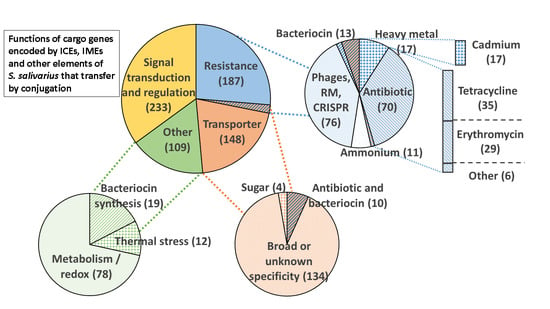
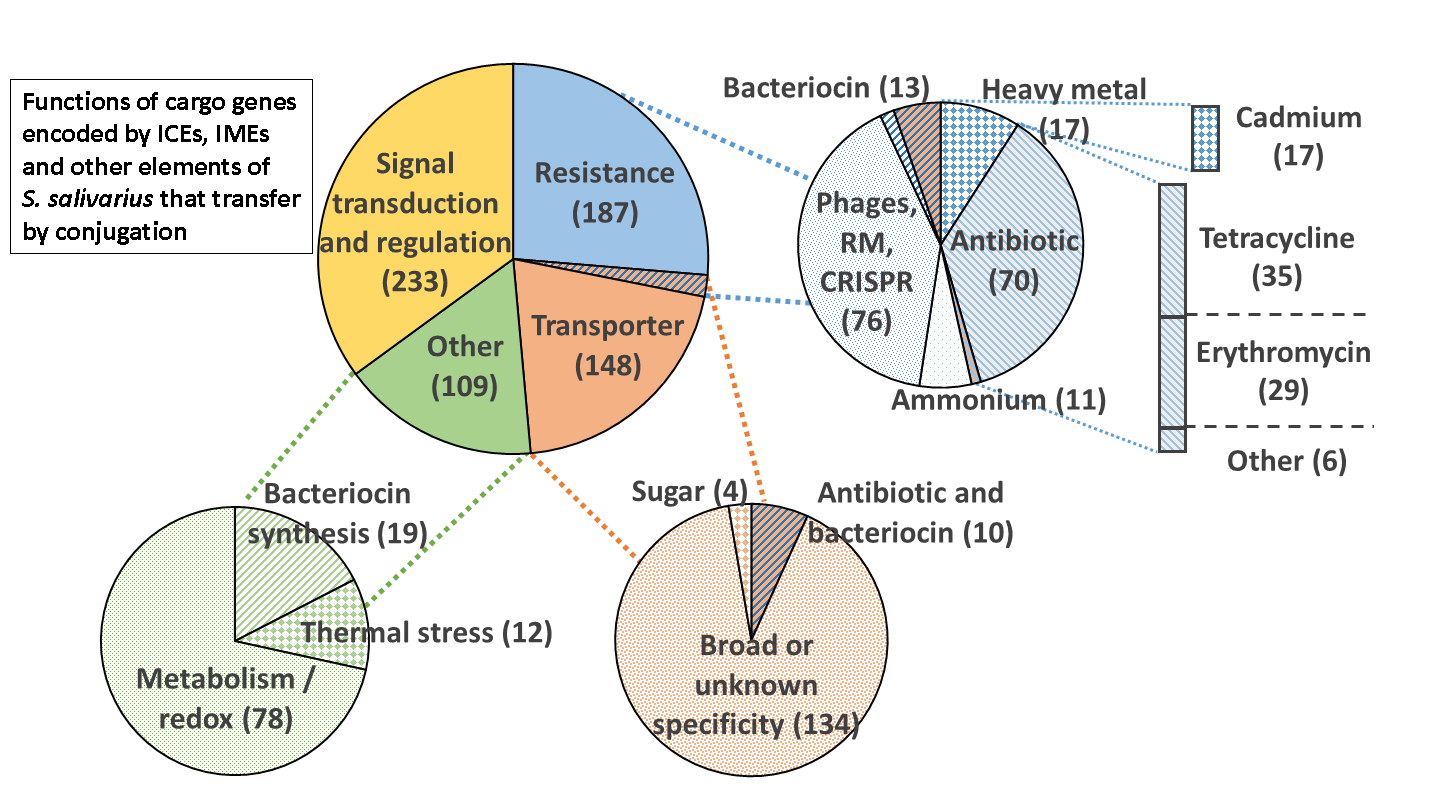
3.4. Function Encoded by the Cargo Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guglielmini, J.; Quintais, L.; Garcillán-Barcia, M.P.; de la Cruz, F.; Rocha, E.P.C. The Repertoire of ICE in Prokaryotes Underscores the Unity, Diversity, and Ubiquity of Conjugation. PLoS Genet. 2011, 7, e1002222. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, X.; Payot, S.; Leblond-Bourget, N.; Guédon, G. Conjugative and mobilizable genomic islands in bacteria: Evolution and diversity. FEMS Microbiol. Rev. 2014, 38, 720–760. [Google Scholar] [CrossRef]
- Guédon, G.; Libante, V.; Coluzzi, C.; Payot, S.; Leblond-Bourget, N. The Obscure World of Integrative and Mobilizable Elements, Highly Widespread Elements that Pirate Bacterial Conjugative Systems. Genes 2017, 8, 337. [Google Scholar] [CrossRef] [PubMed]
- Delavat, F.; Miyazaki, R.; Carraro, N.; Pradervand, N.; van der Meer, J.R. The hidden life of integrative and conjugative elements. FEMS Microbiol. Rev. 2017, 41, 512–537. [Google Scholar] [CrossRef]
- Toussaint, A.; Merlin, C. Mobile Elements as a Combination of Functional Modules. Plasmid 2002, 47, 26–35. [Google Scholar] [CrossRef]
- Burrus, V.; Pavlovic, G.; Decaris, B.; Guédon, G. Conjugative transposons: The tip of the iceberg. Mol. Microbiol. 2002, 46, 601–610. [Google Scholar] [CrossRef]
- Wozniak, R.A.F.; Waldor, M.K. Integrative and conjugative elements: Mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010, 8, 552–563. [Google Scholar] [CrossRef]
- Goessweiner-Mohr, N.; Arends, K.; Keller, W.; Grohmann, E. Conjugative type IV secretion systems in Gram-positive bacteria. Plasmid 2013, 70, 289–302. [Google Scholar] [CrossRef]
- Guglielmini, J.; Néron, B.; Abby, S.S.; Garcillán-Barcia, M.P.; de la Cruz, F.; Rocha, E.P.C. Key components of the eight classes of type IV secretion systems involved in bacterial conjugation or protein secretion. Nucleic Acids Res. 2014, 42, 5715–5727. [Google Scholar] [CrossRef]
- Johnson, C.M.; Grossman, A.D. Integrative and Conjugative Elements (ICEs): What They Do and How They Work. Annu. Rev. Genet. 2015, 49, 577–601. [Google Scholar] [CrossRef] [PubMed]
- Garcillán-Barcia, M.P.; Redondo-Salvo, S.; Vielva, L.; de la Cruz, F. MOBscan: Automated Annotation of MOB Relaxases. In Horizontal Gene Transfer: Methods and Protocols; De la Cruz, F., Ed.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2020; pp. 295–308. [Google Scholar]
- Ambroset, C.; Coluzzi, C.; Guédon, G.; Devignes, M.-D.; Loux, V.; Lacroix, T.; Payot, S.; Leblond-Bourget, N. New Insights into the Classification and Integration Specificity of Streptococcus Integrative Conjugative Elements through Extensive Genome Exploration. Front. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Garcillán-Barcia, M.P.; Cuartas-Lanza, R.; Cuevas, A.; Cruz, F. de la Cis-Acting Relaxases Guarantee Independent Mobilization of MOBQ4 Plasmids. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.; Miguel-Arribas, A.; Abia, D.; Singh, P.K.; Crespo, I.; Gago-Córdoba, C.; Hao, J.A.; Luque-Ortega, J.R.; Alfonso, C.; Wu, L.J.; et al. Discovery of a new family of relaxases in Firmicutes bacteria. PLoS Genet. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Soler, N.; Robert, E.; Chauvot de Beauchêne, I.; Monteiro, P.; Libante, V.; Maigret, B.; Staub, J.; Ritchie, D.W.; Guédon, G.; Payot, S.; et al. Characterization of a relaxase belonging to the MOBT family, a widespread family in Firmicutes mediating the transfer of ICEs. Mob. DNA 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, C.; Guédon, G.; Devignes, M.-D.; Ambroset, C.; Loux, V.; Lacroix, T.; Payot, S.; Leblond-Bourget, N. A Glimpse into the World of Integrative and Mobilizable Elements in Streptococci Reveals an Unexpected Diversity and Novel Families of Mobilization Proteins. Front. Microbiol. 2017, 8, 443. [Google Scholar] [CrossRef]
- Bellanger, X.; Morel, C.; Gonot, F.; Puymege, A.; Decaris, B.; Guédon, G. Site-specific accretion of an integrative conjugative element together with a related genomic island leads to cis mobilization and gene capture. Mol. Microbiol. 2011, 81, 912–925. [Google Scholar] [CrossRef]
- Libante, V.; Nombre, Y.; Coluzzi, C.; Staub, J.; Guédon, G.; Gottschalk, M.; Teatero, S.; Fittipaldi, N.; Leblond-Bourget, N.; Payot, S. Chromosomal Conjugative and Mobilizable Elements in Streptococcus suis: Major Actors in the Spreading of Antimicrobial Resistance and Bacteriocin Synthesis Genes. Pathogens 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef]
- Hegstad, K.; Mylvaganam, H.; Janice, J.; Josefsen, E.; Sivertsen, A.; Skaare, D. Role of Horizontal Gene Transfer in the Development of Multidrug Resistance in Haemophilus influenzae. mSphere 2020, 5, e00969-19. [Google Scholar] [CrossRef]
- Mullany, P.; Allan, E.; Roberts, A.P. Mobile genetic elements in Clostridium difficile and their role in genome function. Res. Microbiol. 2015, 166, 361–367. [Google Scholar] [CrossRef]
- Fernandez-Gutierrez, M.M.; Roosjen, P.P.J.; Ultee, E.; Agelink, M.; Vervoort, J.J.M.; Keijser, B.; Wells, J.M.; Kleerebezem, M. Streptococcus salivarius MS-oral-D6 promotes gingival re-epithelialization in vitro through a secreted serine protease. Sci. Rep. 2017, 7, 11100. [Google Scholar] [CrossRef] [PubMed]
- Couvigny, B.; de Wouters, T.; Kaci, G.; Jacouton, E.; Delorme, C.; Doré, J.; Renault, P.; Blottière, H.M.; Guédon, E.; Lapaque, N. Commensal Streptococcus salivarius Modulates PPARγ Transcriptional Activity in Human Intestinal Epithelial Cells. PLoS ONE 2015, 10, e0125371. [Google Scholar] [CrossRef] [PubMed]
- Kaci, G.; Goudercourt, D.; Dennin, V.; Pot, B.; Doré, J.; Ehrlich, S.D.; Renault, P.; Blottière, H.M.; Daniel, C.; Delorme, C. Anti-Inflammatory Properties of Streptococcus salivarius, a Commensal Bacterium of the Oral Cavity and Digestive Tract. Appl. Environ. Microbiol. 2014, 80, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.; Martin, R.; Walk, S.T.; Young, C.; Grossman, S.; McKean, E.L.; Aronoff, D.M. Clinical and laboratory features of Streptococcus salivarius meningitis: A case report and literature review. Clin. Med. Res. 2012, 10, 15–25. [Google Scholar] [CrossRef]
- Shirokawa, T.; Nakajima, J.; Hirose, K.; Suzuki, H.; Nagaoka, S.; Suzuki, M. Spontaneous meningitis due to Streptococcus salivarius subsp. salivarius: Cross-reaction in an assay with a rapid diagnostic kit that detected Streptococcus pneumoniae antigens. Intern. Med. Tokyo Jpn. 2014, 53, 279–282. [Google Scholar] [CrossRef][Green Version]
- Kitten, T.; Munro, C.L.; Zollar, N.Q.; Lee, S.P.; Patel, R.D. Oral streptococcal bacteremia in hospitalized patients: Taxonomic identification and clinical characterization. J. Clin. Microbiol. 2012, 50, 1039–1042. [Google Scholar] [CrossRef][Green Version]
- Corredoira, J.C.; Alonso, M.P.; García, J.F.; Casariego, E.; Coira, A.; Rodriguez, A.; Pita, J.; Louzao, C.; Pombo, B.; López, M.J.; et al. Clinical characteristics and significance of Streptococcus salivarius bacteremia and Streptococcus bovis bacteremia: A prospective 16-year study. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2005, 24, 250–255. [Google Scholar] [CrossRef]
- Han, X.Y.; Kamana, M.; Rolston, K.V.I. Viridans streptococci isolated by culture from blood of cancer patients: Clinical and microbiologic analysis of 50 cases. J. Clin. Microbiol. 2006, 44, 160–165. [Google Scholar] [CrossRef]
- Delorme, C.; Poyart, C.; Ehrlich, S.D.; Renault, P. Extent of Horizontal Gene Transfer in Evolution of Streptococci of the Salivarius Group. J. Bacteriol. 2007, 189, 1330–1341. [Google Scholar] [CrossRef]
- Delorme, C.; Abraham, A.-L.; Renault, P.; Guédon, E. Genomics of Streptococcus salivarius, a major human commensal. Infect. Genet. Evol. 2015, 33, 381–392. [Google Scholar] [CrossRef]
- Dahmane, N.; Libante, V.; Charron-Bourgoin, F.; Guédon, E.; Guédon, G.; Leblond-Bourget, N.; Payot, S. Diversity of Integrative and Conjugative Elements of Streptococcus salivarius and Their Intra- and Interspecies Transfer. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Chaffanel, F.; Charron-Bourgoin, F.; Libante, V.; Leblond-Bourget, N.; Payot, S. Resistance Genes and Genetic Elements Associated with Antibiotic Resistance in Clinical and Commensal Isolates of Streptococcus salivarius. Appl. Environ. Microbiol. 2015, 81, 4155–4163. [Google Scholar] [CrossRef] [PubMed]
- Guedon, E.; Delorme, C.; Pons, N.; Cruaud, C.; Loux, V.; Couloux, A.; Gautier, C.; Sanchez, N.; Layec, S.; Galleron, N.; et al. Complete Genome Sequence of the Commensal Streptococcus salivarius Strain JIM8777. J. Bacteriol. 2011, 193, 5024–5025. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.-H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Hammami, R.; Zouhir, A.; Le Lay, C.; Ben Hamida, J.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef]
- Flissi, A.; Ricart, E.; Campart, C.; Chevalier, M.; Dufresne, Y.; Michalik, J.; Jacques, P.; Flahaut, C.; Lisacek, F.; Leclère, V.; et al. Norine: Update of the nonribosomal peptide resource. Nucleic Acids Res. 2020, 48, D465–D469. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Roberts, A.P.; Mullany, P. Tn916-like genetic elements: A diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 856–871. [Google Scholar] [CrossRef] [PubMed]
- Brochet, M.; Couvé, E.; Glaser, P.; Guédon, G.; Payot, S. Integrative Conjugative Elements and Related Elements Are Major Contributors to the Genome Diversity of Streptococcus agalactiae. J. Bacteriol. 2008, 190, 6913–6917. [Google Scholar] [CrossRef] [PubMed]
- Guérillot, R.; Siguier, P.; Gourbeyre, E.; Chandler, M.; Glaser, P. The Diversity of Prokaryotic DDE Transposases of the Mutator Superfamily, Insertion Specificity, and Association with Conjugation Machineries. Genome Biol. Evol. 2014, 6, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Carraro, N.; Libante, V.; Morel, C.; Decaris, B.; Charron-Bourgoin, F.; Leblond, P.; Guédon, G. Differential regulation of two closely related integrative and conjugative elements from Streptococcus thermophilus. BMC Microbiol. 2011, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Libante, V.; Sarica, N.; Mohamad Ali, A.; Gapp, C.; Oussalah, A.; Guédon, G.; Leblond-Bourget, N.; Payot, S. Mobilization of IMEs integrated in the oriT of ICEs involves their own relaxase belonging to the Rep-trans family of proteins. Genes 2020, in press. [Google Scholar]
- Rawls, K.S.; Yacovone, S.K.; Maupin-Furlow, J.A. GlpR Represses Fructose and Glucose Metabolic Enzymes at the Level of Transcription in the Haloarchaeon Haloferax volcanii. J. Bacteriol. 2010, 192, 6251–6260. [Google Scholar] [CrossRef]
- Chen, Y.-Y.M.; Feng, C.W.; Chiu, C.F.; Burne, R.A. cadDX Operon of Streptococcus salivarius 57.I. Appl. Environ. Microbiol. 2008, 74, 1642–1645. [Google Scholar] [CrossRef]
- Stock, A.M.; Robinson, V.L.; Goudreau, P.N. Two-Component Signal Transduction. Annu. Rev. Biochem. 2000, 35, 183–215. [Google Scholar] [CrossRef]
- Martin, B.; Granadel, C.; Campo, N.; Hénard, V.; Prudhomme, M.; Claverys, J.-P. Expression and maintenance of ComD–ComE, the two-component signal-transduction system that controls competence of Streptococcus pneumoniae. Mol. Microbiol. 2010, 75, 1513–1528. [Google Scholar] [CrossRef]
- Zhulin, I.B.; Taylor, B.L.; Dixon, R. PAS domain S-boxes in Archaea, Bacteria and sensors for oxygen and redox. Trends Biochem. Sci. 1997, 22, 331–333. [Google Scholar] [CrossRef]
- Ponting, C.P.; Aravind, L. PAS: A multifunctional domain family comes to light. Curr. Biol. CB 1997, 7, R674–R677. [Google Scholar] [CrossRef]
- Malbruny, B.; Werno, A.M.; Murdoch, D.R.; Leclercq, R.; Cattoir, V. Cross-Resistance to Lincosamides, Streptogramins A, and Pleuromutilins Due to the lsa (C) Gene in Streptococcus agalactiae UCN70. Antimicrob. Agents Chemother. 2011, 55, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Kathariou, S. Transposon-induced mutants of Listeria monocytogenes incapable of growth at low temperature (4°C). FEMS Microbiol. Lett. 1994, 121, 287–291. [Google Scholar] [CrossRef]
- Puymège, A.; Bertin, S.; Guédon, G.; Payot, S. Analysis of Streptococcus agalactiae pan-genome for prevalence, diversity and functionality of integrative and conjugative or mobilizable elements integrated in the tRNALys CTT gene. Mol. Genet. Genomics 2015, 290, 1727–1740. [Google Scholar] [CrossRef]
- Van Eijk, E.; Anvar, S.; Browne, H.P.; Leung, W.; Frank, J.; Schmitz, A.M.; Roberts, A.P.; Smits, W. Complete genome sequence of the Clostridium difficile laboratory strain 630Δerm reveals differences from strain 630, including translocation of the mobile element CTn5. BMC Genom. 2015, 16, 31. [Google Scholar] [CrossRef]
- Hochhut, B.; Beaber, J.W.; Woodgate, R.; Waldor, M.K. Formation of Chromosomal Tandem Arrays of the SXT Element and R391, Two Conjugative Chromosomally Integrating Elements That Share an Attachment Site. J. Bacteriol. 2001, 183, 1124–1132. [Google Scholar] [CrossRef]
- Possoz, C.; Ribard, C.; Gagnat, J.; Pernodet, J.-L.; Guérineau, M. The integrative element pSAM2 from Streptomyces: Kinetics and mode of conjugal transfer. Mol. Microbiol. 2001, 42, 159–166. [Google Scholar] [CrossRef]
- Song, L.; Jiang, Y.; Zhang, X. Chronology and pattern of integration of tandem genomic islands associated with the tmRNA gene in Escherichia coli and Salmonella enterica. Chin. Sci. Bull. 2011, 56, 3836–3843. [Google Scholar] [CrossRef]
- Chapleau, M.; Guertin, J.F.; Farrokhi, A.; Lerat, S.; Burrus, V.; Beaulieu, C. Identification of genetic and environmental factors stimulating excision from Streptomyces scabiei chromosome of the toxicogenic region responsible for pathogenicity. Mol. Plant Pathol. 2016, 17, 501–509. [Google Scholar] [CrossRef]
- Wang, P.; Zeng, Z.; Wang, W.; Wen, Z.; Li, J.; Wang, X. Dissemination and loss of a biofilm-related genomic island in marine Pseudoalteromonas mediated by integrative and conjugative elements. Environ. Microbiol. 2017, 19, 4620–4637. [Google Scholar] [CrossRef] [PubMed]
- León-Sampedro, R.; Fernández-de-Bobadilla, M.D.; San Millán, Á.; Baquero, F.; Coque, T.M. Transfer dynamics of Tn6648, a composite integrative conjugative element generated by tandem accretion of Tn5801 and Tn6647 in Enterococcus faecalis. J. Antimicrob. Chemother. 2019, 74, 2517–2523. [Google Scholar] [CrossRef] [PubMed]
- Goeders, N.; Chai, R.; Chen, B.; Day, A.; Salmond, G.P.C. Structure, Evolution, and Functions of Bacterial Type III Toxin-Antitoxin Systems. Toxins 2016, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II Toxin-Antitoxin Systems: Evolution and Revolutions. J. Bacteriol. 2020, 202, e00763-19. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Mruk, I.; Kobayashi, I. To be or not to be: Regulation of restriction–modification systems and other toxin–antitoxin systems. Nucleic Acids Res. 2014, 42, 70–86. [Google Scholar] [CrossRef]
- Kobayashi, I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001, 29, 3742–3756. [Google Scholar] [CrossRef]
- Burrus, V.; Bontemps, C.; Decaris, B.; Guédon, G. Characterization of a Novel Type II Restriction-Modification System, Sth368I, Encoded by the Integrative Element ICESt1 of Streptococcus thermophilusCNRZ368. Appl. Environ. Microbiol. 2001, 67, 1522–1528. [Google Scholar] [CrossRef]
- Chaidez, C.; Lopez, J.; Castro-del Campo, N. Quaternary ammonium compounds: An alternative disinfection method for fresh produce wash water. J. Water Health 2007, 5, 329–333. [Google Scholar] [CrossRef]
- Järup, L.; Åkesson, A. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharmacol. 2009, 238, 201–208. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Integrase Type (Number of IMEs/dIMEs) | Relaxase Families (Number of Relaxases) | CP Families (Number of CPs) | IME Superfamilies 1 |
|---|---|---|---|
| Tyrosine (n = 155) | Rel_PF02486_2 (n = 65) | none (n = 65) | IME_PF02486 (IME_class_1) |
| Rel _PF02486_4 (n = 4) | TcpA_2 (n = 4) | ||
| Rel _PF02486_5 (n = 21) | none (n = 22) | ||
| Rel _PF02486_6 (n = 22) | none (n = 22) | ||
| Rel _PF01719 (n = 28) | TcpA_12 (n = 28) | IME_PF01719 (IME_class_2) | |
| Rel_PF01719-PF0910_4 (n = 3) Rel_PF01719-PF0910_5 (n = 4) | TcpA_6 (n = 1) TcpA_14 (n = 1) none (n = 1) TcpA_12 (n = 4) | IME_ PF01719-PF0910 (IME_class_4) | |
| Rel_PHA00330_1 (n = 3) | TcpA_15 (n = 1) none (n = 2) | IME_PHA00330 (IME_class_3) | |
| Rel_PHA00330_2 (n = 2) | TcpA_8 (n = 2) | ||
| Rel_PHA00330_3 (n = 1) | TcpA_13 (n = 1) | ||
| Rel_PF13814 (n = 1) | none (n = 1) | IME_PF13814 (IME_class_8) | |
| Serine (n = 12) | Rel_PF13814 (n = 12) | VirD4 (n = 12) |
| Structure of Composite Regions | Prevalence |
|---|---|
| CIME-IME-target gene | 7 |
| microCIME-ICE-target gene | 6 |
| CIME-ICE-target gene | 4 |
| IME-IME-target gene | 3 |
| CIME-IME-target gene | 3 |
| microCIME-IME-target gene | 2 |
| 2 microCIMEs-dICE-target gene | 2 |
| IME-ICE-target gene | 2 |
| ICE-IME-target gene | 2 |
| CIME-IME-IME-target gene | 2 |
| microCIME-CIME-ICE-target gene | 1 |
| CIME-dICE-target gene | 1 |
| CIME-ICE-IME-target gene | 1 |
| MGI-IME-target gene | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lao, J.; Guédon, G.; Lacroix, T.; Charron-Bourgoin, F.; Libante, V.; Loux, V.; Chiapello, H.; Payot, S.; Leblond-Bourget, N. Abundance, Diversity and Role of ICEs and IMEs in the Adaptation of Streptococcus salivarius to the Environment. Genes 2020, 11, 999. https://doi.org/10.3390/genes11090999
Lao J, Guédon G, Lacroix T, Charron-Bourgoin F, Libante V, Loux V, Chiapello H, Payot S, Leblond-Bourget N. Abundance, Diversity and Role of ICEs and IMEs in the Adaptation of Streptococcus salivarius to the Environment. Genes. 2020; 11(9):999. https://doi.org/10.3390/genes11090999
Chicago/Turabian StyleLao, Julie, Gérard Guédon, Thomas Lacroix, Florence Charron-Bourgoin, Virginie Libante, Valentin Loux, Hélène Chiapello, Sophie Payot, and Nathalie Leblond-Bourget. 2020. "Abundance, Diversity and Role of ICEs and IMEs in the Adaptation of Streptococcus salivarius to the Environment" Genes 11, no. 9: 999. https://doi.org/10.3390/genes11090999
APA StyleLao, J., Guédon, G., Lacroix, T., Charron-Bourgoin, F., Libante, V., Loux, V., Chiapello, H., Payot, S., & Leblond-Bourget, N. (2020). Abundance, Diversity and Role of ICEs and IMEs in the Adaptation of Streptococcus salivarius to the Environment. Genes, 11(9), 999. https://doi.org/10.3390/genes11090999