
Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cohort Presentation
2.2. DNA Extraction and Sanger Sequencing
2.3. Relative Allele Frequency Calculation
2.4. In Silico Analysis of Variants
3. Results
3.1. Patients Characteristics
3.2. Novel ALMS1 pathogenic variants
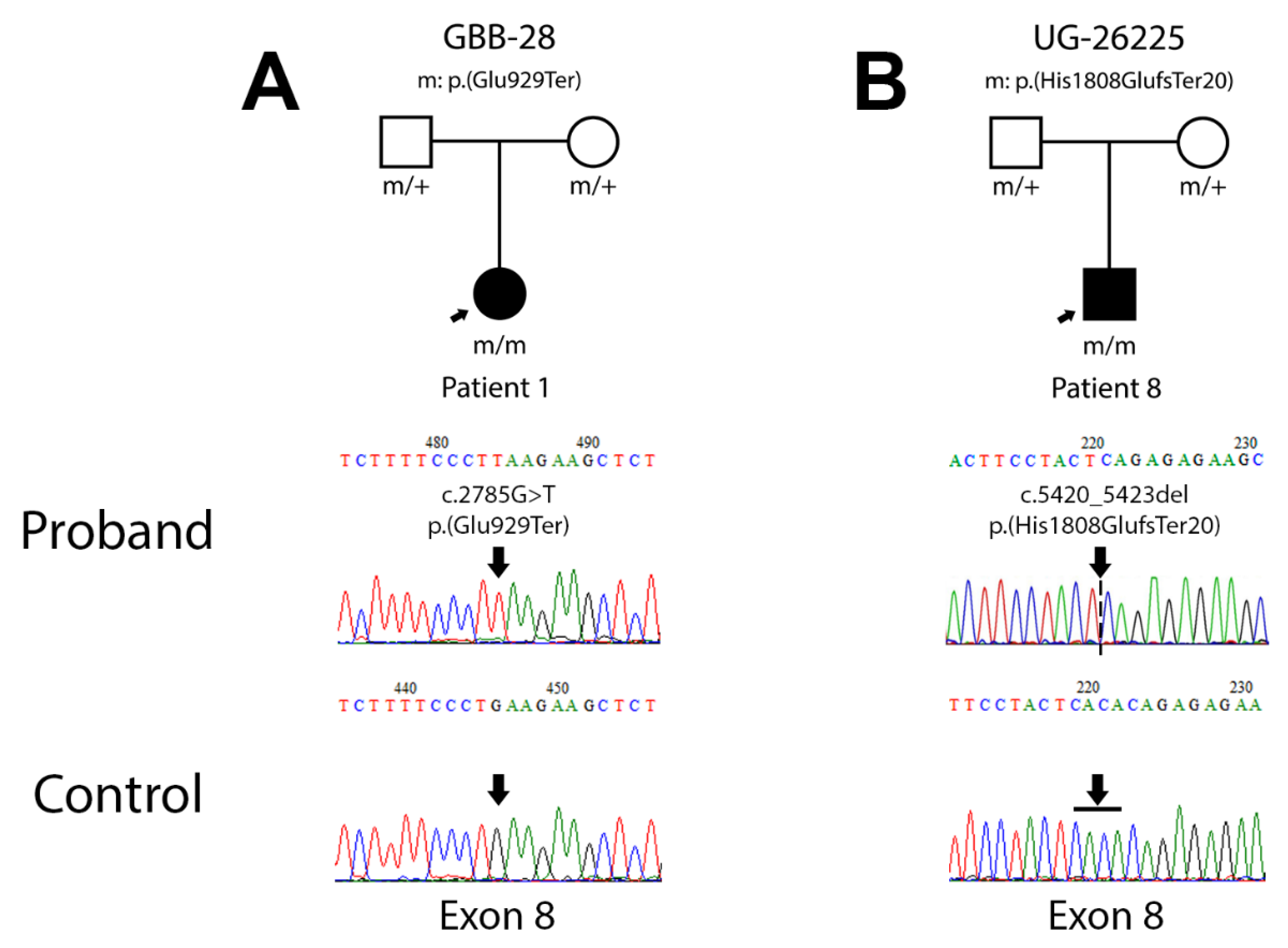
3.2.1. Patient 1 (Family GBB-28)
3.2.2. Patient 8 (Family UG-26225)
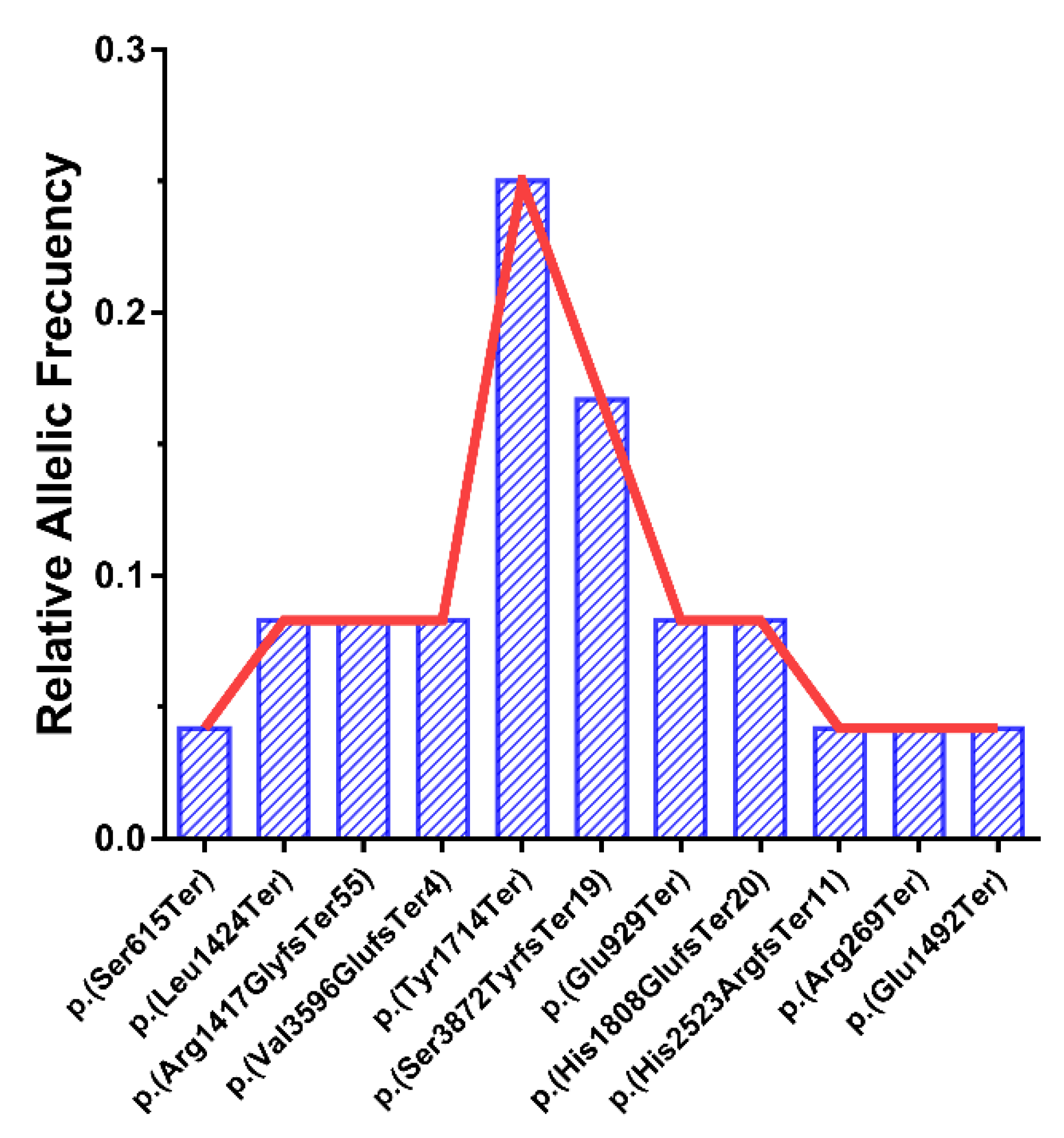
3.3. Relative Allele Frequencies
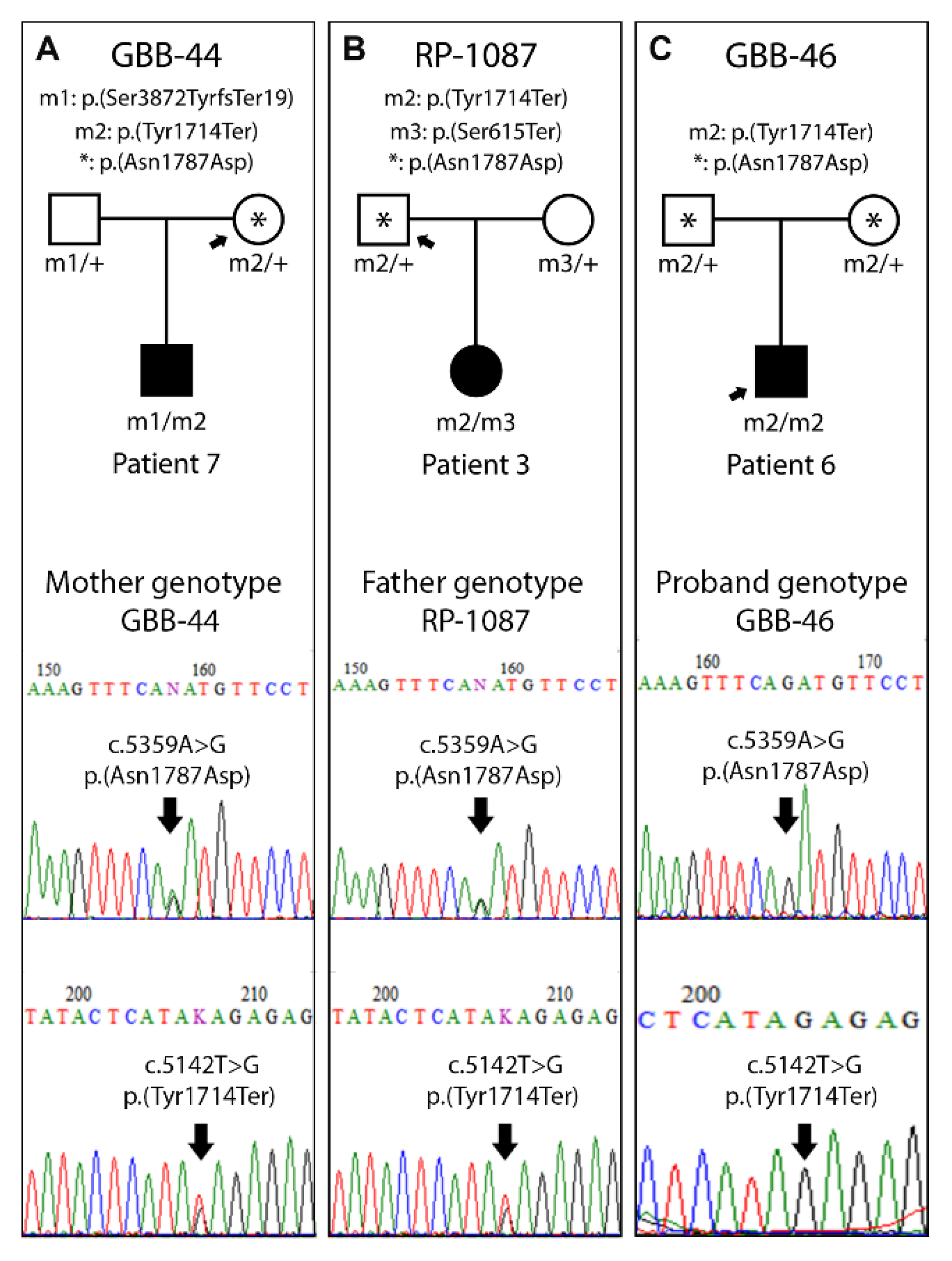
3.4. Segregation Study
3.5. Haplogroup Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marshall, J.D.; Ludman, M.D.; Shea, S.E.; Salisbury, S.R.; Willi, S.M.; LaRoche, R.G.; Nishina, P.M. Genealogy, natural history, and phenotype of Alström syndrome in a large Acadian kindred and three additional families. Am. J. Med. Genet. 1997, 73, 150–161. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Safieh, L.A.; Alkuraya, H.; Al-Rajhi, A.; Shamseldin, H.; Hashem, M.; Alzahrani, F.; Khan, A.O.; Alqahtani, F.; Rahbeeni, Z.; et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol. Vis. 2009, 15, 2464–2469. [Google Scholar] [PubMed]
- Ozantürk, A.; Marshall, J.D.; Collin, G.B.; Düzenli, S.; Marshall, R.P.; Candan, Ş.; Tos, T.; Esen, İ.; Taşkesen, M.; Çayır, A.; et al. The phenotypic and molecular genetic spectrum of Alström syndrome in 44 Turkish kindreds and a literature review of Alström syndrome in Turkey. J. Hum. Genet. 2014, 60, 1. [Google Scholar] [CrossRef]
- Marshall, J.D.; Muller, J.; Collin, G.B.; Milan, G.; Kingsmore, S.F.; Dinwiddie, D.; Farrow, E.G.; Miller, N.A.; Favaretto, F.; Maffei, P.; et al. Alström Syndrome: Mutation Spectrum of ALMS1. Hum. Mutat. 2015, 36, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alström syndrome: Genetics and clinical overview. Curr. Genomics 2011, 12, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro-Gallego, T.; Cortón, M.; Ayuso, C.; Baiget, M.; Valverde, D. Molecular approach in the study of Alström syndrome: Analysis of ten Spanish families. Mol. Vis. 2012, 18, 1794–1802. [Google Scholar]
- Sanchez-Navarro, I.; da Silva, L.R.J.; Blanco-Kelly, F.; Zurita, O.; Sanchez-Bolivar, N.; Villaverde, C.; Lopez-Molina, M.I.; Garcia-Sandoval, B.; Tahsin-Swafiri, S.; Minguez, P.; et al. Combining targeted panel-based resequencing and copy-number variation analysis for the diagnosis of inherited syndromic retinopathies and associated ciliopathies. Sci. Rep. 2018, 8, 5285. [Google Scholar] [CrossRef] [PubMed]
- Tahani, N.; Maffei, P.; Dollfus, H.; Paisey, R.; Valverde, D.; Milan, G.; Han, J.C.; Favaretto, F.; Madathil, S.C.; Dawson, C.; et al. Consensus clinical management guidelines for Alström syndrome. Orphanet J. Rare Dis. 2020, 15, 253. [Google Scholar] [CrossRef]
- Marshall, J.D.; Beck, S.; Maffei, P.; Naggert, J.K. Alström Syndrome. Eur. J. Hum. Genet. 2007, 15, 1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, G.B.; Marshall, J.D.; Ikeda, A.; So, W.V.; Russell-Eggitt, I.; Maffei, P.; Beck, S.; Boerkoel, C.F.; Sicolo, N.; Martin, M.; et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat. Genet. 2002, 31, 74. [Google Scholar] [CrossRef]
- Hearn, T.; Renforth, G.L.; Spalluto, C.; Hanley, N.A.; Piper, K.; Brickwood, S.; White, C.; Connolly, V.; Taylor, J.F.N.; Russell-Eggitt, I.; et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat. Genet. 2002, 31, 79. [Google Scholar] [CrossRef] [PubMed]
- Collin, G.B.; Cyr, E.; Bronson, R.; Marshall, J.D.; Gifford, E.J.; Hicks, W.; Murray, S.A.; Zheng, Q.Y.; Smith, R.S.; Nishina, P.M.; et al. Alms1-disrupted mice recapitulate human Alström syndrome. Hum. Mol. Genet. 2005, 14, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Hearn, T.; Spalluto, C.; Phillips, V.J.; Renforth, G.L.; Copin, N.; Hanley, N.A.; Wilson, D.I. Subcellular Localization of ALMS1 Supports Involvement of Centrosome and Basal Body Dysfunction in the Pathogenesis of Obesity, Insulin Resistance, and Type 2 Diabetes. Diabetes 2005, 54, 1581–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braune, K.; Volkmer, I.; Staege, M.S. Characterization of Alstrom Syndrome 1 (ALMS1) Transcript Variants in Hodgkin Lymphoma Cells. PLoS ONE 2017, 12, e0170694. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Sabir, A.; Fulton, P.; Zatyka, M.; Williams, D.; Hardy, C.; Milan, G.; Favaretto, F.; Yu-Wai-Man, P.; Rohayem, J.; et al. Monogenic diabetes syndromes: Locus-specific databases for Alström, Wolfram, and Thiamine-responsive megaloblastic anemia. Hum. Mutat. 2017, 38, 764–777. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Cao, M.; Li, Z.; Chen, X.; Patenia, C.; Gore, A.; Abboud, E.B.; Al-Rajhi, A.A.; Lewis, R.A.; et al. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Hum. Mutat. 2011, 32, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, S.; Yoshitake, K.; Akahori, M.; Hayashi, T.; Furuno, M.; Nishino, J.; Ikeo, K.; Tsuneoka, H.; Iwata, T. Whole-exome sequencing identifies a novel ALMS1 mutation (p.Q2051X) in two Japanese brothers with Alström syndrome. Mol. Vis. 2013, 19, 2393–2406. [Google Scholar]
- Long, P.A.; Evans, J.M.; Olson, T.M. Exome sequencing establishes diagnosis of Alström syndrome in an infant presenting with non-syndromic dilated cardiomyopathy. Am. J. Med. Genet. A 2015, 167A, 886–890. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.D.; Hinman, E.G.; Collin, G.B.; Beck, S.; Cerqueira, R.; Maffei, P.; Milan, G.; Zhang, W.; Wilson, D.I.; Hearn, T.; et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum. Mutat. 2007, 28, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Lazar, C.H.; Kimchi, A.; Namburi, P.; Mutsuddi, M.; Zelinger, L.; Beryozkin, A.; Ben-Simhon, S.; Obolensky, A.; Ben-Neriah, Z.; Argov, Z.; et al. Nonsyndromic Early-Onset Cone-Rod Dystrophy and Limb-Girdle Muscular Dystrophy in a Consanguineous Israeli Family are Caused by Two Independent yet Linked Mutations in ALMS1 and DYSF. Hum. Mutat. 2015, 36, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeldt, L.B.; Biswas, S.; Madeoy, J.; Connelly, C.F.; Schadt, E.E.; Akey, J.M. Population genomic analysis of ALMS1 in humans reveals a surprisingly complex evolutionary history. Mol. Biol. Evol. 2009, 26, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Groenendael, S.; Giacovazzi, L.; Davison, F.; Holtkemper, O.; Huang, Z.; Wang, Q.; Parkinson, K.; Barrett, T.; Geberhiwot, T. High quality, patient centred and coordinated care for Alstrom syndrome: A model of care for an ultra-rare disease. Orphanet J. Rare Dis. 2015, 10, 149. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Allele 1 | Allele 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient | Family | Reference | ALMS1 Pathogenic Variant 1 c.DNA | Exon | ALMS1 Pathogenic Variant 1 Protein | ALMS1 Pathogenic Variant 2 c.DNA | Exon | ALMS1 Pathogenic Variant 2 Protein | Genotype Status |
| 1 | GBB-28 | This study | c.2785G>T | 8 | p.(Glu929Ter) | c.2785G>T | 8 | p.(Glu929Ter) | Homozygous |
| 2 | RP-1232 | [7] | c.4249del | 8 | p.(Arg1417GlyfsTer55) | c.4249del | 8 | p.(Arg1417GlyfsTer55) | Homozygous |
| 3 | RP-1087 | [6] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.1844C>G | 8 | p.(Ser615Ter) | Heterozygous |
| 4 | GAS-37 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.4271T>G | 8 | p.(Leu1424Ter) | Heterozygous |
| 5 | GAS-37 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.4271T>G | 8 | p.(Leu1424Ter) | Heterozygous |
| 6 | GBB-46 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.5142T>G | 8 | p.(Tyr1714Ter) | Homozygous |
| 7 | GBB-44 | Allele 1 [6]; Allele 2 [4] | c.5142T>G | 8 | p.(Tyr1714Ter) | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | Heterozygous |
| 8 | UG-26225 | This study | c.5420_5423del | 8 | p.(His1808GlufsTer20) | c.5420_5423del | 8 | p.(His1808GlufsTer20) | Homozygous |
| 9 | RP-2186 | [4] | c.7568_7569del | 9 | p.(His2523ArgfsTer11) | c.4474G>T | 8 | p.(Glu1492Ter) | Heterozygous |
| 10 | RP-793 | [19] | c.10787_10788del | 16 | p.(Val3596GlufsTer4) | c.10787_10788del | 16 | p.(Val3596GlufsTer4) | Homozygous |
| 11 | GBB-45 | [4] | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | Homozygous |
| 12 | RP-2177 | Allele1 [4]; Allele 2 [20] | c.11615_11616del | 17 | p.(Ser3872TyrfsTer19) | c.805C>T | 5 | p.(Arg269Ter) | Heterozygous |
| Patient | Family | Sex | Age (Years) | Vision (History of Nystagmus in Infancy/Childhood, Legal Blindness, Cone and Rod Dystrophy by ERG) | Obesity and/or Insulin Resistance and/or T2D | History of DCM/CHF | Hearing Loss | Hepatic Dysfunction | Renal Failure | Short Stature | Males: Hypogonadism; Females: Irregular Menses and/or Hyperandrogenism | Thyroid Disorders | Predicted Protein Change |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GBB-28 | F | 13 | x | x | x | - | - | - | - | - | - | p.(Glu929Ter)/p.(Glu929Ter) |
| 2 | RP-1232 | F | 27 | x | x | - | x | - | x | - | x | x | p.(Arg1417GlyfsTer55)/p.(Arg1417GlyfsTer55) |
| 3 | RP-1087 | F | 42 | x | x | - | x | x | x | x | x | - | p.(Tyr1714Ter)/p.(Ser615Ter) |
| 4 | GAS-37 | F | 21 | x | x | x | x | x | - | x | x | x | p.(Tyr1714Ter)/p.(Leu1424Ter) |
| 5 | GAS-37 | M | 26 | x | x | - | x | x | - | x | x | x | p.(Tyr1714Ter)/p.(Leu1424Ter) |
| 6 | GBB-46 | M | 23 | x | x | - | x | x | - | x | - | x | p.(Tyr1714Ter)/p.(Tyr1714Ter) |
| 7 | GBB-44 | M | 18 | x | x | x | x | x | - | - | - | - | p.(Tyr1714Ter)/p.(Ser3872TyrfsTer19) |
| 8 | UG-26225 | M | 3 | x | x | x | - | - | - | - | - | x | p.(His1808GlufsTer20)/p.(His1808GlufsTer20) |
| 9 | RP-2186 | M | 9 | x | x | x | - | - | - | - | - | - | p.(His2523ArgfsTer11)/p.(Glu1492Ter) |
| 10 | RP-793 | F | 11 | x | x | - | x | x | - | x | - | x | p.(Val3596GlufsTer4)/p.(Val3596GlufsTer4) |
| 12 | RP-2177 | F | 49 | x | - | x | x | - | - | - | - | - | p.(Ser3872TyrfsTer19)/p.(Arg269Ter) |
| Patient | Family | ALMS1 Pathogenic Variant c.DNA | Exon | ALMS1 Pathogenic Variant Protein | PolyPhen2 | MutPred-LOF | SIFT | PROVEAN | ACMG |
|---|---|---|---|---|---|---|---|---|---|
| 1 | GBB-28 | c.2785G>T | 8 | p.(Glu929Ter) | - | 0.432 | 0 | −3.376 | Pathogenic |
| 8 | UG-26225 | c.5420_5423del | 8 | p.(His1808GlufsTer20) | 0.852 | 0.422 | 0,05 | −2.720 | Pathogenic |
| Predicted Protein Change | SNPs | Exon | GBB-44 | RP-1087 | GBB-46 | Common Allele |
|---|---|---|---|---|---|---|
| p.(Phe730=) | rs7598901 | 8 | T/T | T/T | T/T | T |
| p.(Gly1415Val) | rs6546837 | 8 | G/G | C/G | G/G | G |
| p.(Ile1876Val) | rs6546838 | 8 | A/A | G/A | A/A | A |
| p.(Ser2112Arg) | rs6724782 | 8 | T/T | A/T | T/T | T |
| p.(Arg2285Leu) | rs6546839 | 8 | G/G | C/G | G/G | G |
| p.(Arg2827Ser) | rs2056486 | 10 | G/G | G/G | G/G | G |
| p.(Asn2857Ser) | rs10193972 | 10 | A/A | G/A | A/A | A |
| p.(Asn1787Asp) - | rs45608038 | 8 | A/G | A/G | G/G | G |
| p.(Tyr1714Ter) * | rs772136379 | 8 | T/G | T/G | G/G | G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bea-Mascato, B.; Solarat, C.; Perea-Romero, I.; Jaijo, T.; Blanco-Kelly, F.; Millán, J.M.; Ayuso, C.; Valverde, D. Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients. Genes 2021, 12, 282. https://doi.org/10.3390/genes12020282
Bea-Mascato B, Solarat C, Perea-Romero I, Jaijo T, Blanco-Kelly F, Millán JM, Ayuso C, Valverde D. Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients. Genes. 2021; 12(2):282. https://doi.org/10.3390/genes12020282
Chicago/Turabian StyleBea-Mascato, Brais, Carlos Solarat, Irene Perea-Romero, Teresa Jaijo, Fiona Blanco-Kelly, José M. Millán, Carmen Ayuso, and Diana Valverde. 2021. "Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients" Genes 12, no. 2: 282. https://doi.org/10.3390/genes12020282
APA StyleBea-Mascato, B., Solarat, C., Perea-Romero, I., Jaijo, T., Blanco-Kelly, F., Millán, J. M., Ayuso, C., & Valverde, D. (2021). Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients. Genes, 12(2), 282. https://doi.org/10.3390/genes12020282