
Human Mitochondrial Control Region and mtGenome: Design and Forensic Validation of NGS Multiplexes, Sequencing and Analytical Software

Abstract
:1. Introduction
2. Materials and Methods
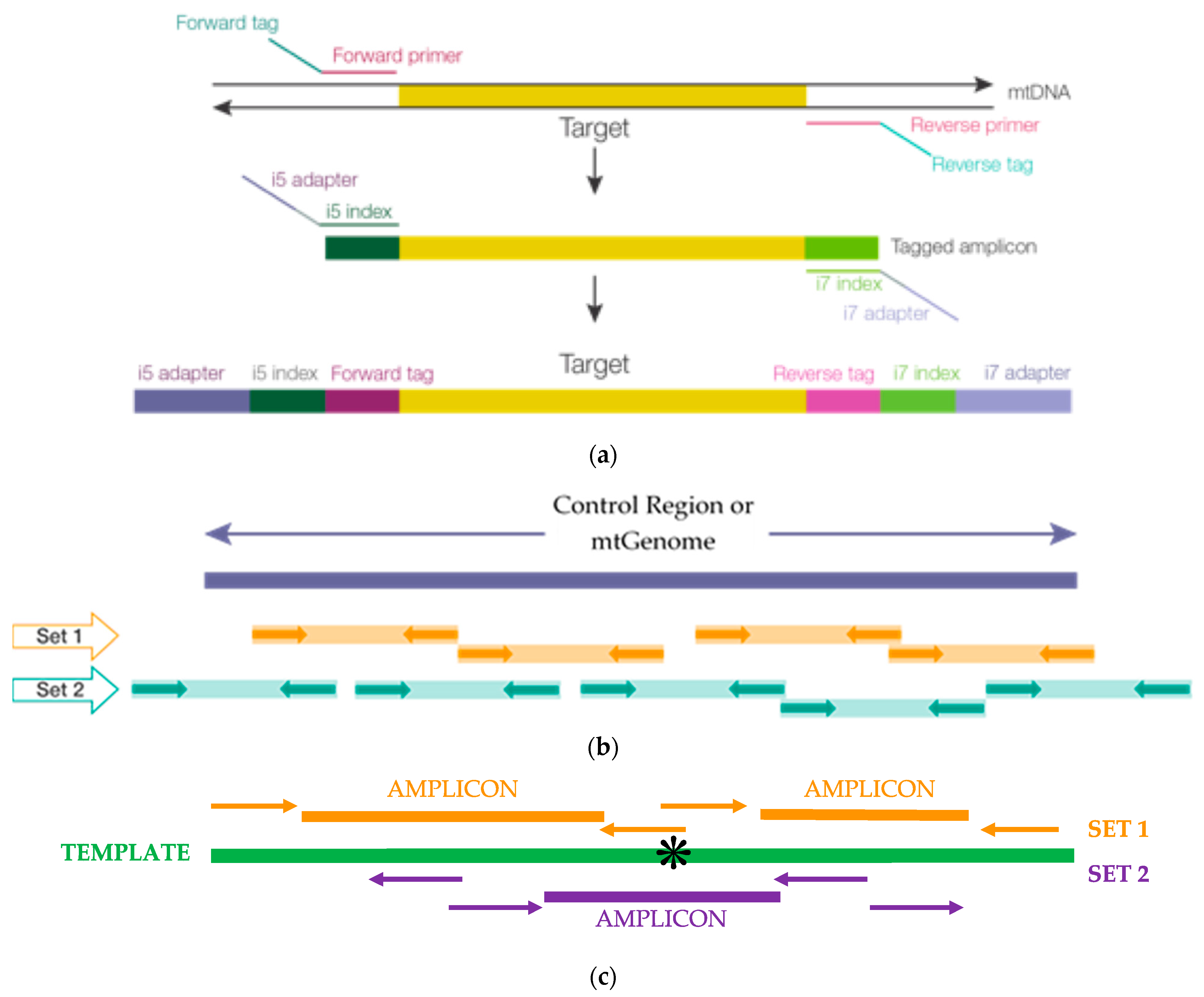
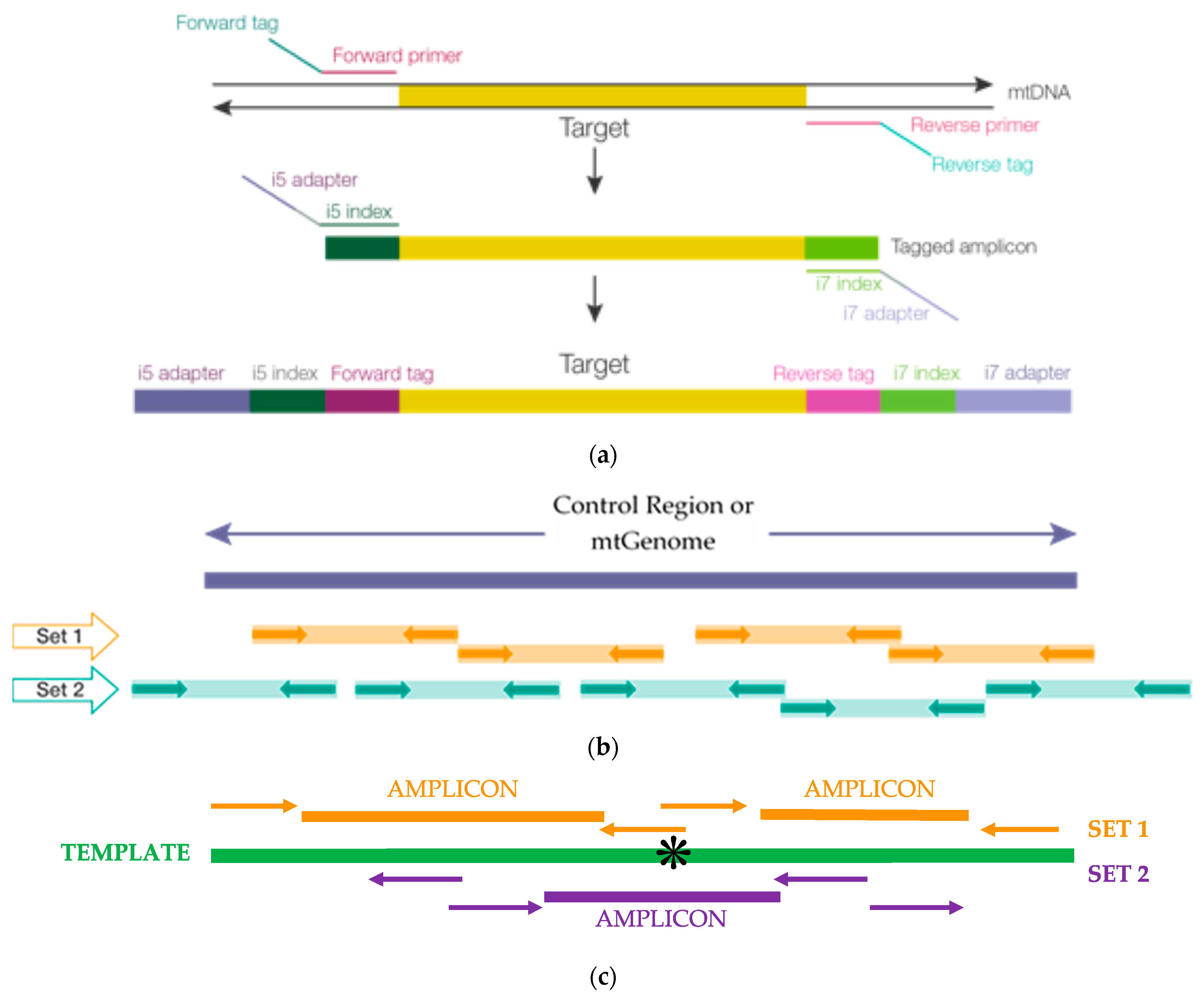
2.1. Primer Design and Placement, Population Studies: mtDNA Control Region and Mitochondrial Genome (mtGenome) Multiplexes
2.2. ForenSeq Positive Control, Human DNA Samples, Mock Casework Samples
2.3. Sensitivity and Mixture DNA Studies
2.4. Repeatability and Reproducibility Studies
2.5. Orthogonal Haplotyping for Concordance Studies
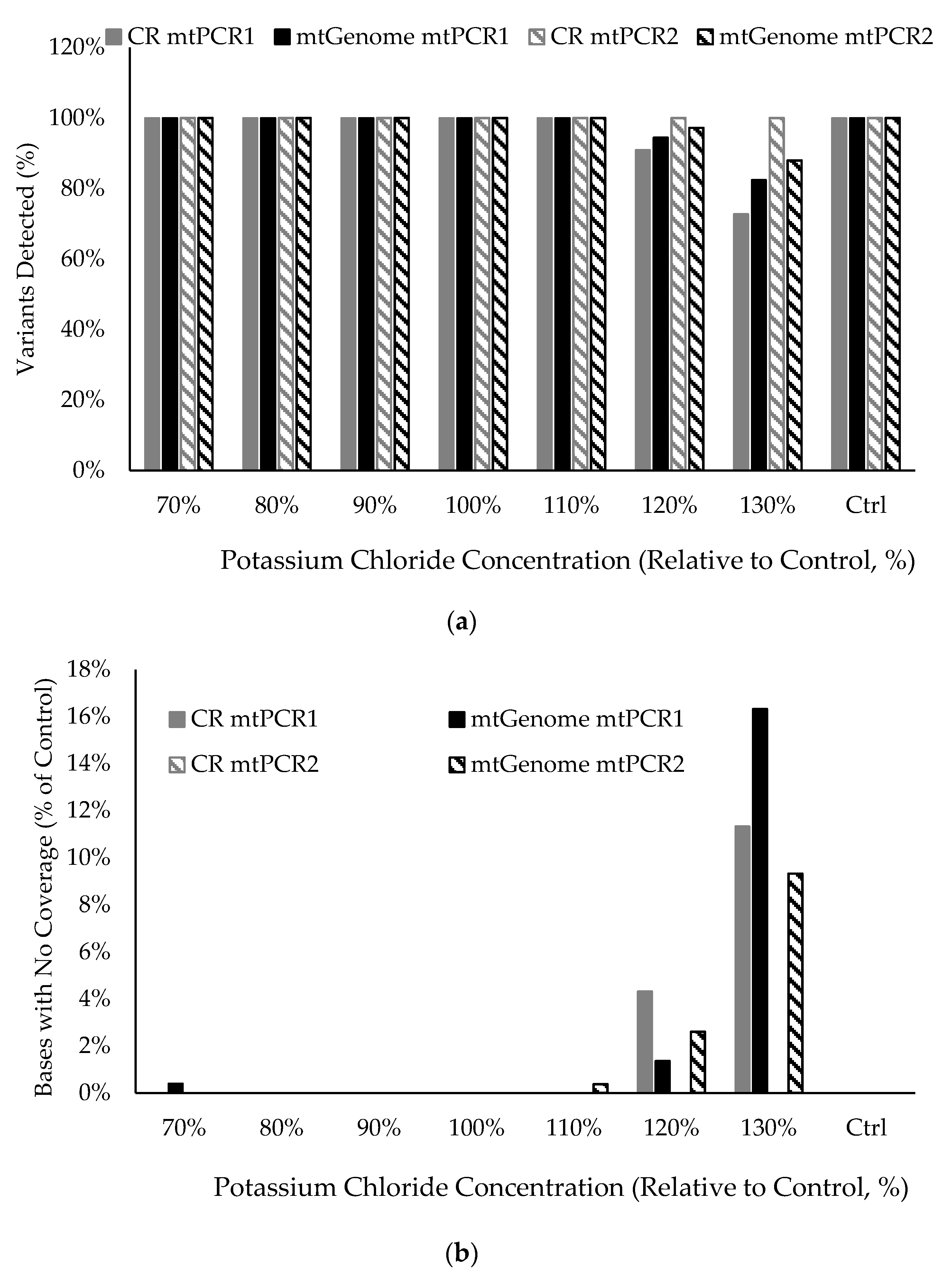
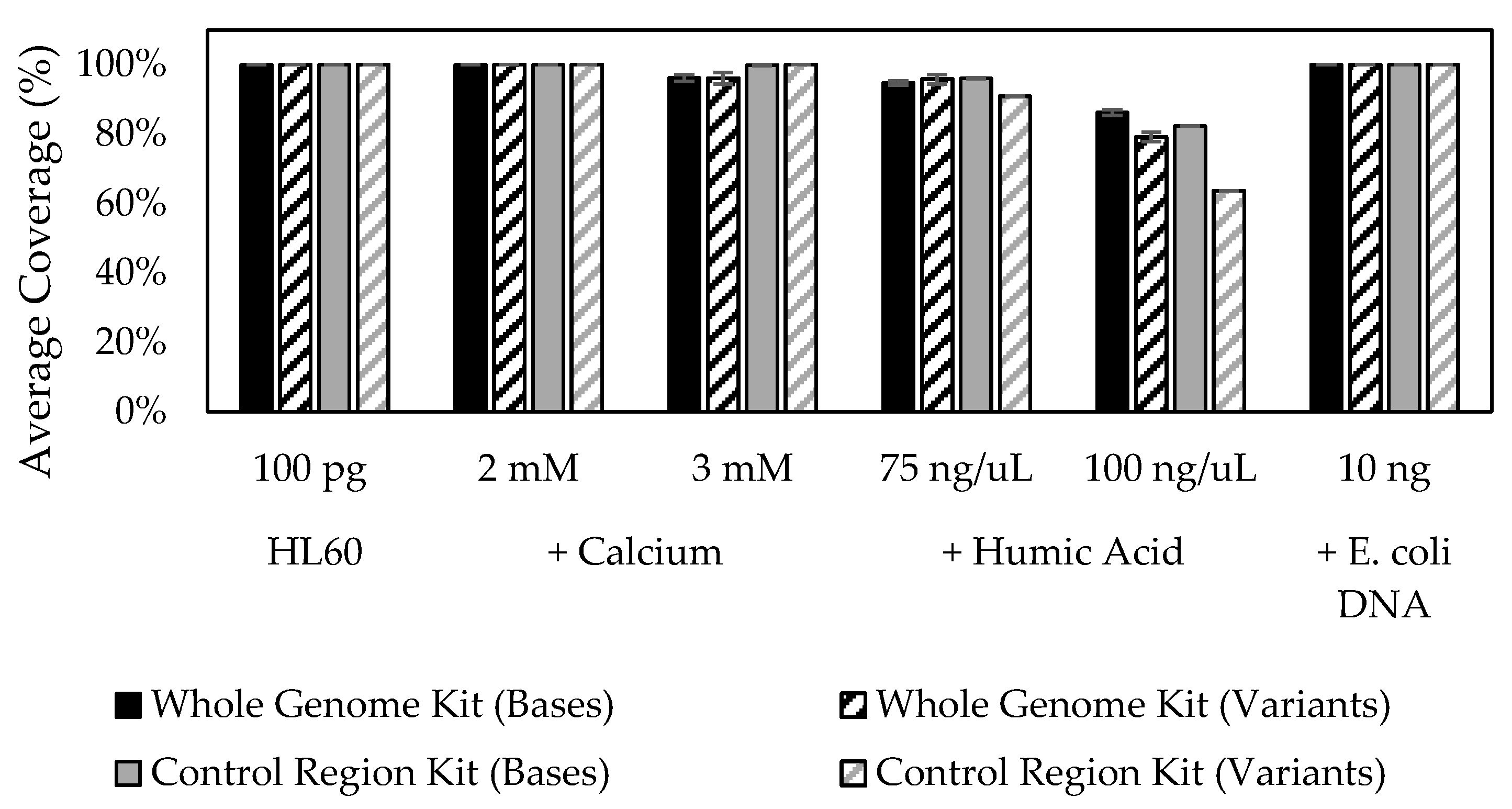
2.6. Stability Studies
2.7. Library Preparation
2.8. MiSeq FGx Sequencing
2.9. NGS Sample Multiplexing and Carryover Assessment
2.10. Contamination and Crosstalk Studies
2.11. PCR-Based Studies
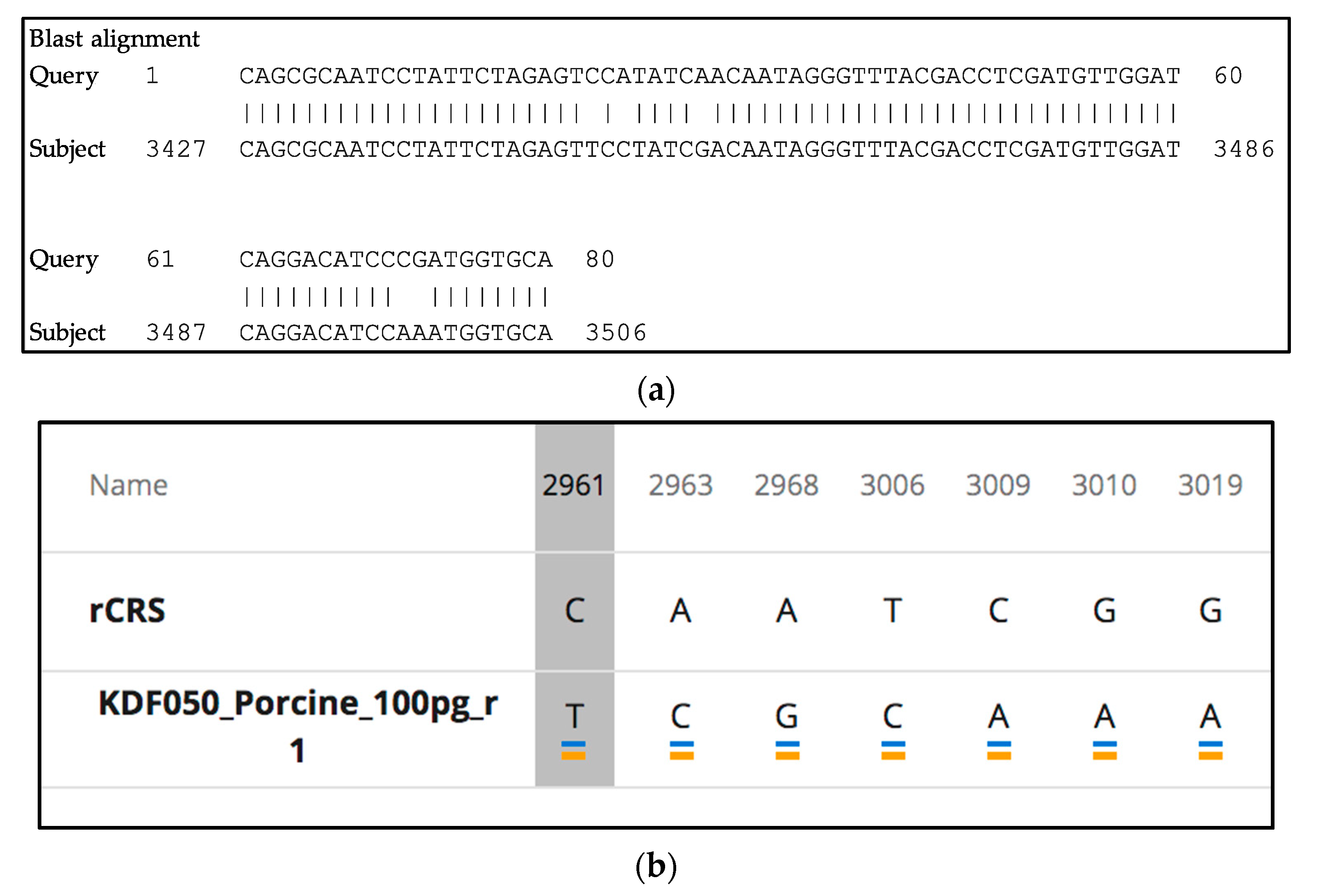
2.12. Species-Specificity Studies
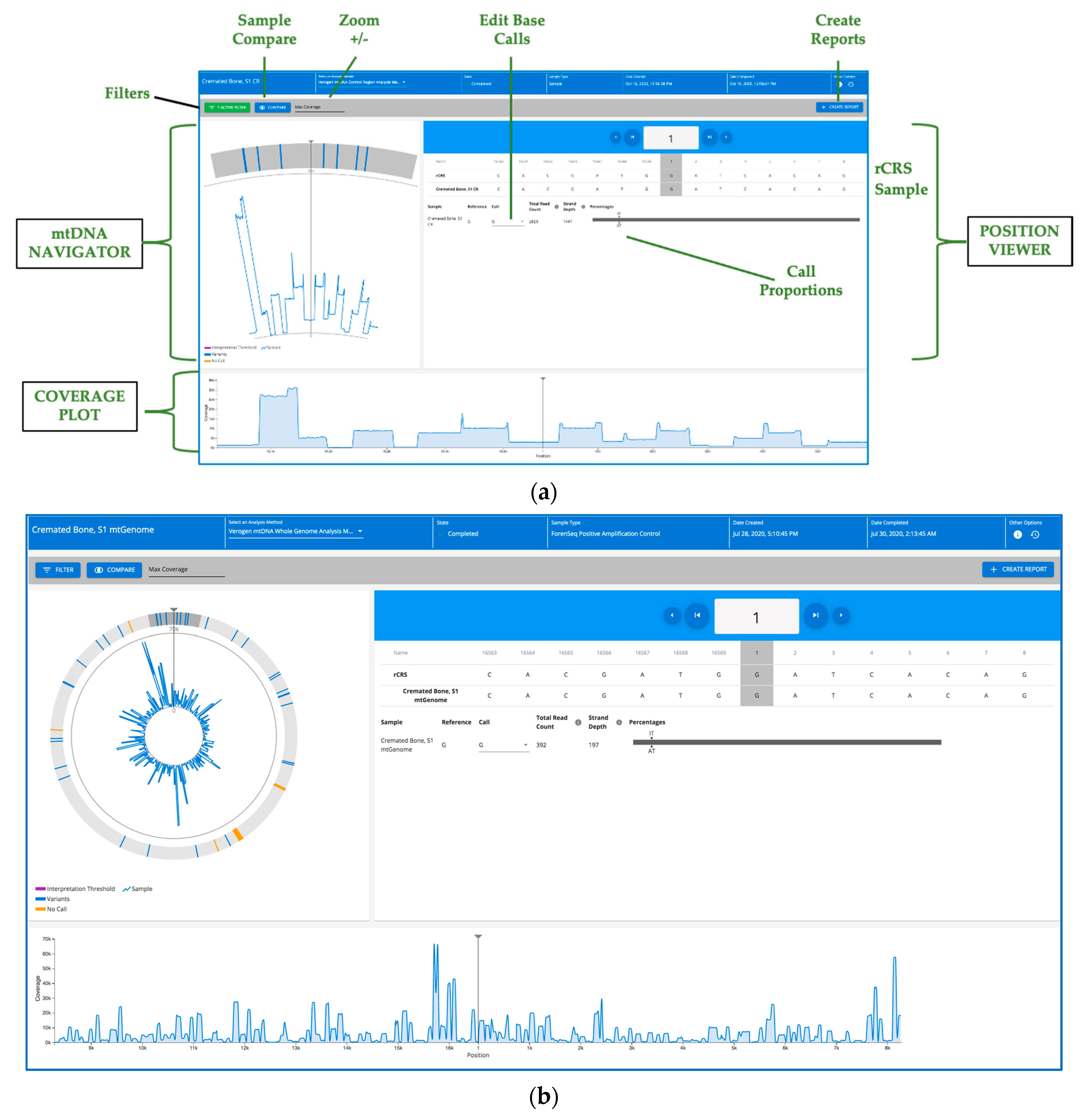
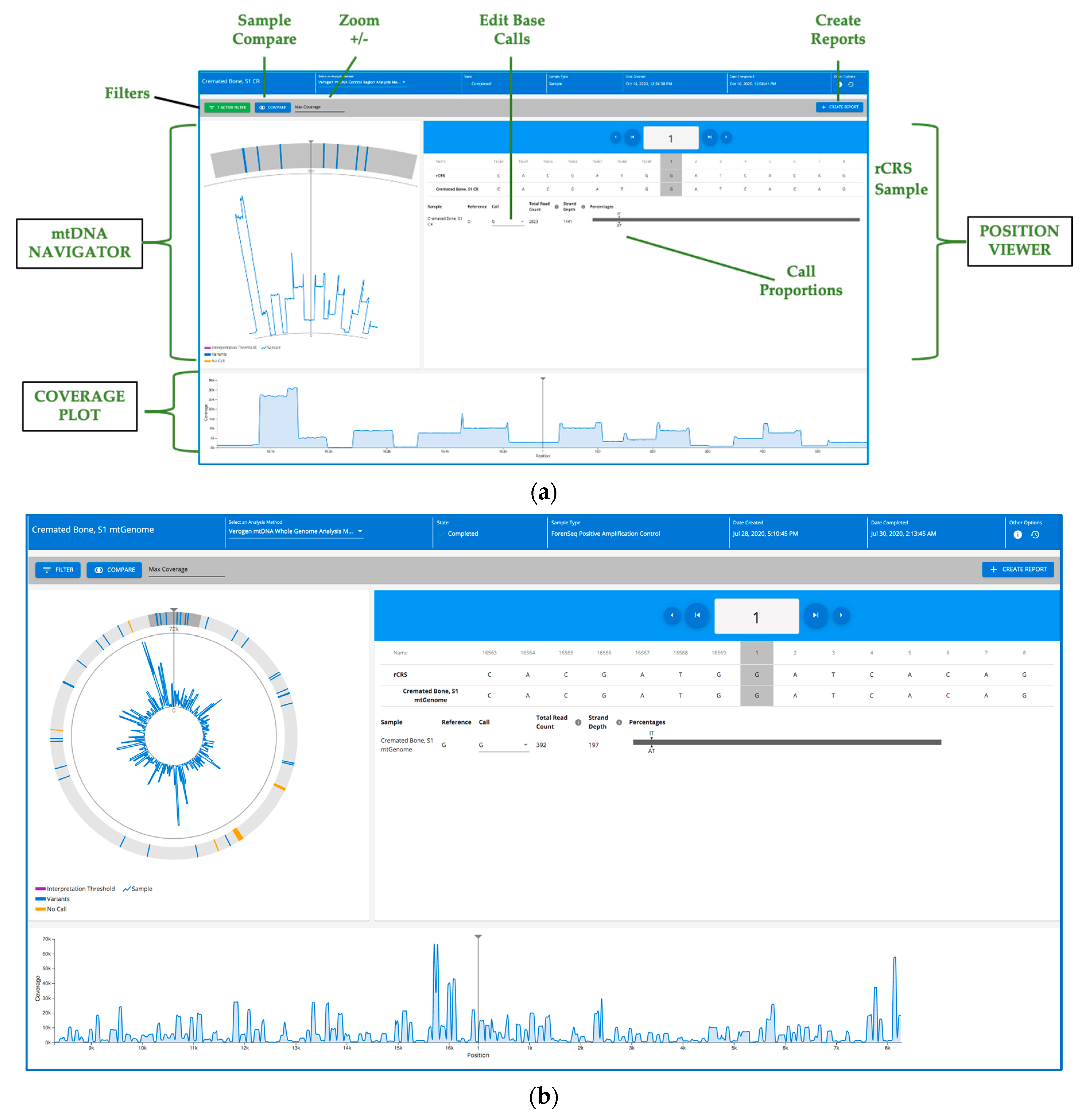
2.13. Secondary and Tertiary Data Analysis
3. Results
3.1. Mock Casework Samples, CR Concordance between Multiplexes
3.1.1. Dental and Bone Samples
3.1.2. Buccal Samples and Rootless Hair Shafts
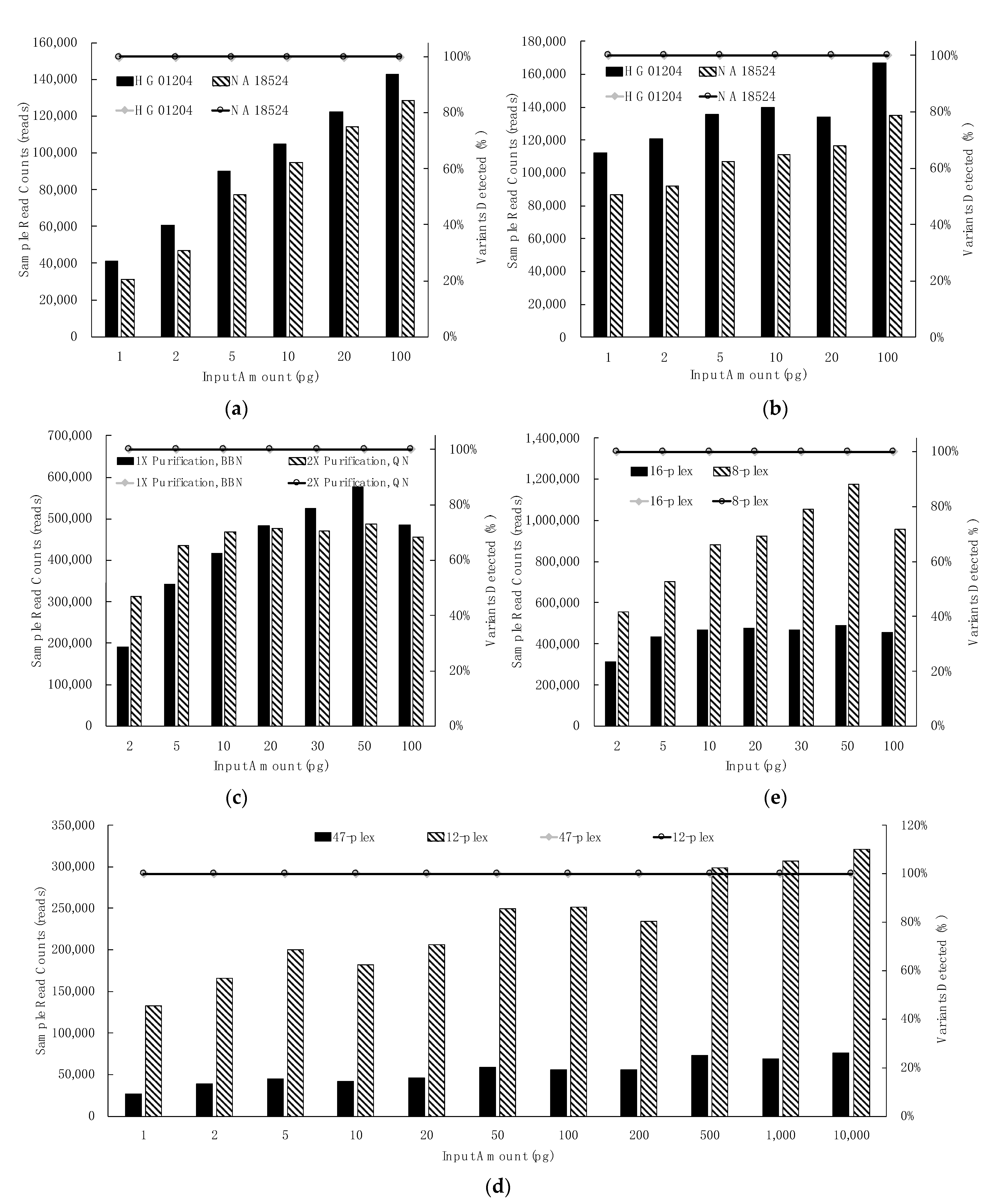
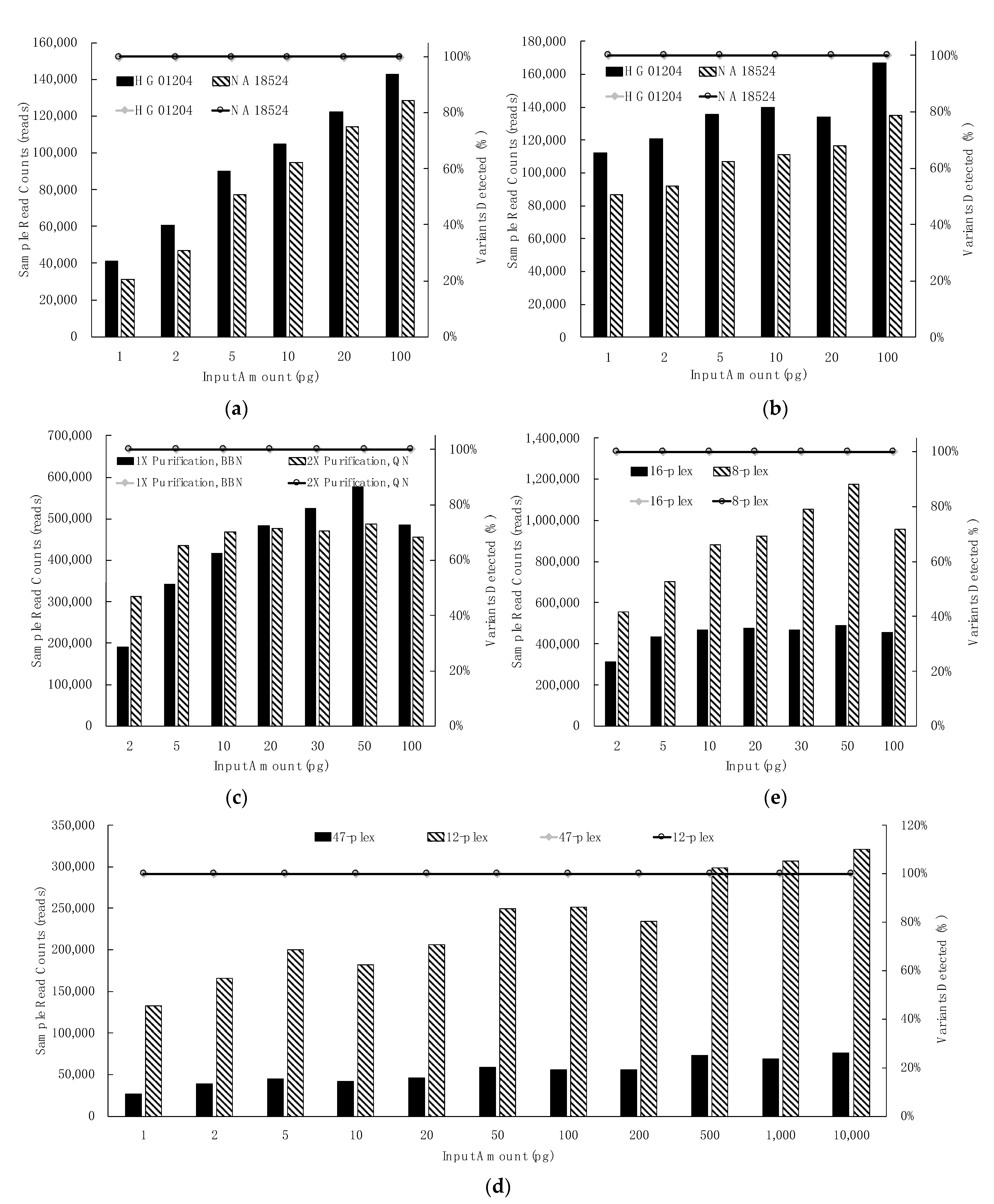
3.2. Sensitivity Studies
3.2.1. Control Region Multiplex: Dilution Series, Library Purification & Library Normalization, Sample Plexity
3.2.2. mtGenome Multiplex: Dilution Series, Library Purification and Normalization, Sample Plexity
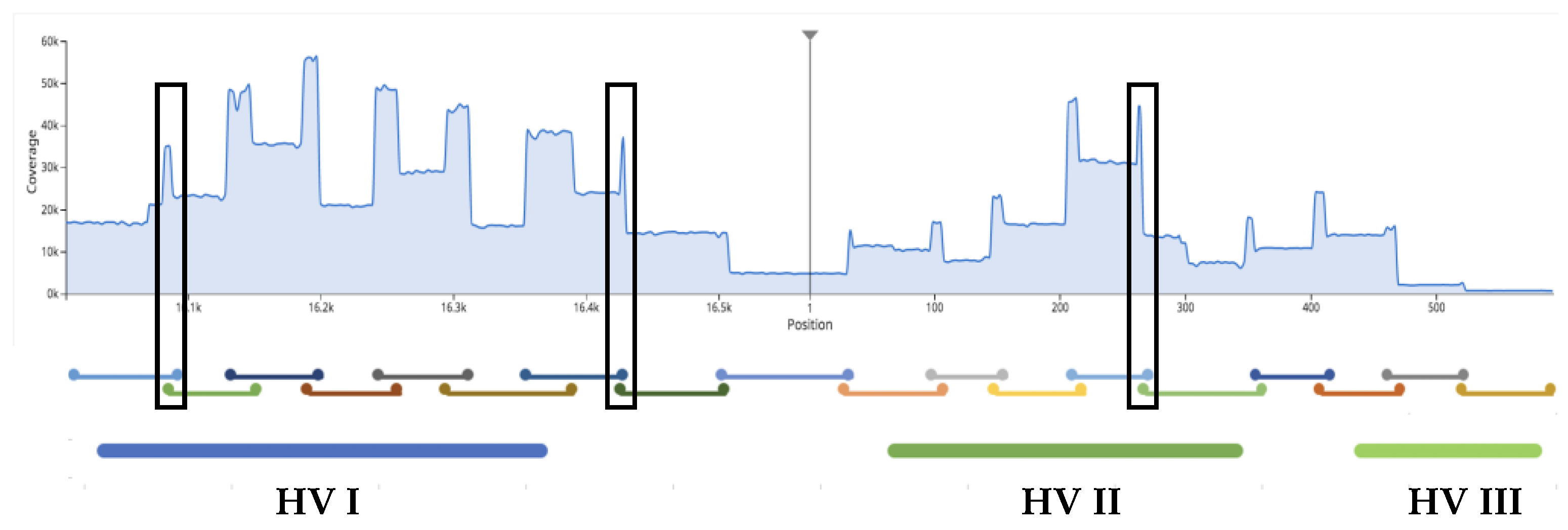
3.2.3. Extent of Sample Multiplexing in Sequencing: Depth of Coverage (DoC)
3.3. Mixture Studies
3.4. Reproducibility and Repeatability: Precision, Accuracy (Concordance), Average Coverage
3.5. Additional Concordance Studies
3.5.1. Control Region Concordance Between ForenSeq Multiplexes
3.5.2. Concordance and Orthogonal Methods, Haplogroup Assignments, Population Studies
3.6. Population Analyses and Studies
3.7. Contamination Assessment
3.7.1. Exogenous DNA
3.7.2. Signal Crosstalk
3.7.3. Sample Carryover between Runs
3.8. PCR-Based Studies
3.8.1. Reaction Conditions
3.8.2. Potential for Differential/Preferential Amplification Among Amplicons
3.8.3. Effects of Amplicon Multiplexing
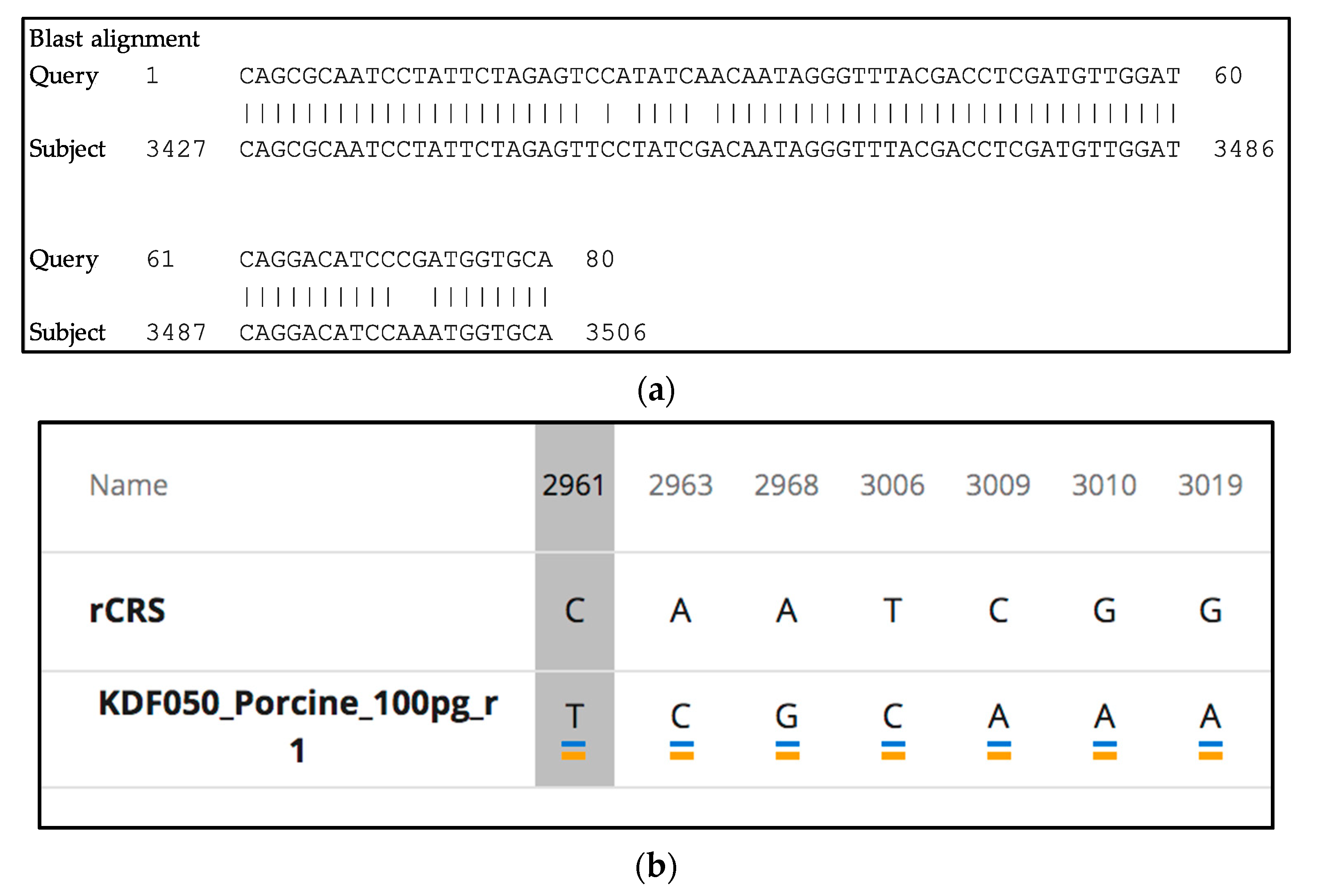
3.9. Species-Specificity
3.9.1. Control Region in the two ForenSeq mtDNA Multiplexes: Species-Specificity
3.9.2. mtGenome Multiplex: Species-Specificity
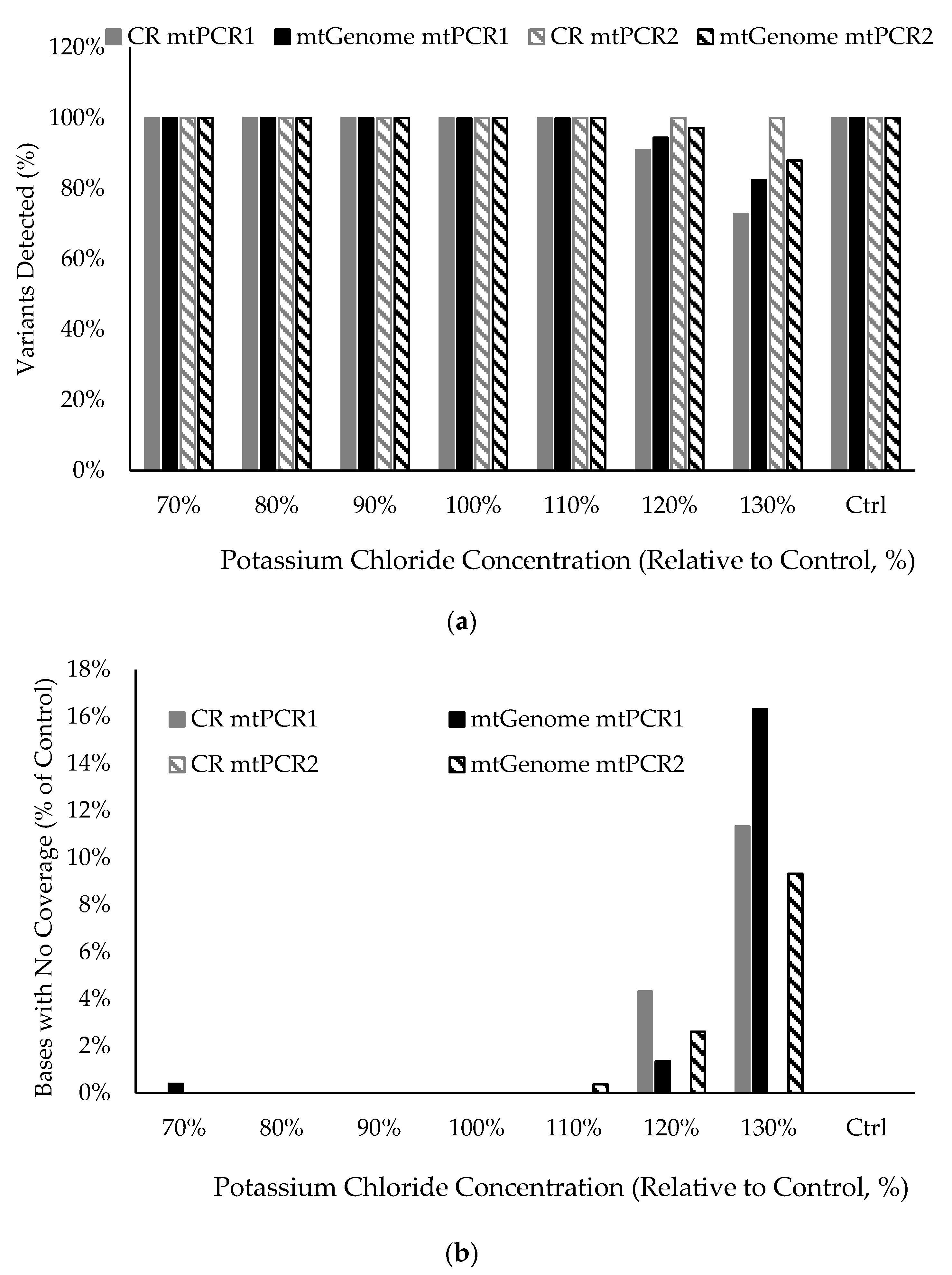
3.10. Stability in the Presence of Inhibitory Substances
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scientific Working Group on DNA Analysis Methods. Validation Guidelines for DNA Analysis Methods. 2016. Available online: https://1ecb9588-ea6f-4feb-971a-73265dbf079c.filesusr.com/ugd/4344b0_813b241e8944497e99b9c45b163b76bd.pdf (accessed on 23 July 2019).
- Federal Bureau of Investigation. Quality Assurance Standards for Forensic DNA Testing Laboratories. 1 July 2020. Available online: https://www.fbi.gov/file-repository/quality-assurance-standards-for-forensic-dna-testing-laboratories.pdf/view (accessed on 1 September 2020).
- Bender, K.; Schneider, P.M.; Rittner, C. Application of mtDNA sequence analysis in forensic casework for the identification of human remains. Forensic Sci. Int. 2000, 113, 103–107. [Google Scholar] [CrossRef]
- Wilson, I.J.; Weale, M.E.; Balding, D.J. Inferences from DNA data: Population histories, evolutionary processes and forensic match probabilities. J. Royal Stat. Soc. Ser. A Stat. Soc. 2003, 166, 155–188. [Google Scholar] [CrossRef]
- Wilson, M.R.; DiZinno, J.A.; Polanskey, D.; Replogle, J.; Budowle, B. Validation of mitochondrial DNA sequencing for forensic casework analysis. Int. J. Leg. Med. 1995, 108, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.M.; Parsons, T.J. Mitochondrial DNA Sequence Analysis—Validation and Use for Forensic Casework. Forensic Sci. Rev. 1999, 11, 21–50. [Google Scholar] [PubMed]
- Almeida, M.; Betancor, E.; Fregel, R.; Suárez, N.M.; Pestano, J. Efficient DNA extraction from hair shafts. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e319–e320. [Google Scholar] [CrossRef]
- Butler, J.M.; Levin, B.C. Forensic applications of mitochondrial DNA. Trends Biotechnol. 1998, 16, 158–162. [Google Scholar] [CrossRef]
- Buckleton, J.S.; Krawczak, M.; Weir, B.S. The interpretation of lineage markers in forensic DNA testing. Forensic Sci. Int. Genet. 2011. [Google Scholar] [CrossRef] [Green Version]
- Fondevila, M.; Phillips, C.; Naveran, N.; Fernandez, L.; Cerezo, M.; Salas, A.; Carracedo, Á.; Lareu, M.V. Case report: Identification of skeletal remains using short-amplicon marker analysis of severely degraded DNA extracted from a decomposed and charred femur. Forensic Sci. Int. Genet. 2008. [Google Scholar] [CrossRef]
- Gill, P.; Ivanov, P.L.; Kimpton, C.; Piercy, R.; Benson, N.; Tully, G.; Evett, I.; Hagelberg, E.; Sullivan, K. Identification of the remains of the romanov family by DNA analysis. Nat. Genet. 1994. [Google Scholar] [CrossRef]
- McNevin, D.; Wilson-Wilde, L.; Robertson, J.; Kyd, J.; Lennard, C. Short tandem repeat (STR) genotyping of keratinised hair: Part 1. Review of current status and knowledge gaps. Forensic Sci. Int. 2005, 153, 237–246. [Google Scholar] [CrossRef]
- McNevin, D.; Wilson-Wilde, L.; Robertson, J.; Kyd, J.; Lennard, C. Short tandem repeat (STR) genotyping of keratinised hair Part 2. An optimised genomic DNA extraction procedure reveals donor dependence of STR profiles. Forensic Sci. Int. 2005, 153, 247–259. [Google Scholar] [CrossRef]
- Tasker, E.; LaRue, B.; Beherec, C.; Gangitano, D.; Hughes-Stamm, S. Analysis of DNA from post-blast pipe bomb fragments for identification and determination of ancestry. Forensic Sci. Int. Genet. 2017, 28, 195–202. [Google Scholar] [CrossRef]
- Houck, M.M.; Budowle, B. Correlation of Microscopic and Mitochondrial DNA Hair Comparisons. J. Forensic Sci. 2002. [Google Scholar] [CrossRef]
- Parson, W.; Strobl, C.; Huber, G.; Zimmermann, B.; Gomes, S.M.; Souto, L.; Fendt, L.; Delport, R.; Langit, R.; Wootton, S.; et al. Evaluation of next generation mtGenome sequencing using the Ion Torrent Personal Genome Machine (PGM). Forensic Sci. Int. Genet. 2013, 7, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Berglund, E.C.; Kiialainen, A.; Syvänen, A.C. Next-generation sequencing technologies and applications for human genetic history and forensics. Investig. Genet. 2011, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- McElhoe, J.A.; Holland, M.M.; Makova, K.D.; Su, M.S.W.; Paul, I.M.; Baker, C.H.; Faith, S.A.; Young, B. Development and assessment of an optimized next-generation DNA sequencing approach for the mtgenome using the Illumina MiSeq. Forensic Sci. Int. Genet. 2014, 13, 20–29. [Google Scholar] [CrossRef] [Green Version]
- King, J.L.; LaRue, B.L.; Novroski, N.M.; Stoljarova, M.; Seo, S.B.; Zeng, X.; Warshauer, D.H.; Davis, C.P.; Parson, W.; Sajantila, A. High-quality and high-throughput massively parallel sequencing of the human mitochondrial genome using the Illumina MiSeq. Forensic Sci. Int. Genet. 2014, 12, 128–135. [Google Scholar] [CrossRef]
- Peck, M.A.; Brandhagen, M.D.; Marshall, C.; Diegoli, T.M.; Irwin, J.A.; Sturk-Andreaggi, K. Concordance and reproducibility of a next generation mtGenome sequencing method for high-quality samples using the Illumina MiSeq. Forensic Sci. Int. Genet. 2016, 24, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Oliver, R.S.; Barritt-Ross, S.; McMahon, T.P. Performance evaluation of a mitogenome capture and Illumina sequencing protocol using non-probative, case-type skeletal samples: Implications for the use of a positive control in a next-generation sequencing procedure. Forensic Sci. Int. Genet. 2017, 31, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Ring, J.D.; Sturk-Andreaggi, K.; Peck, M.A.; Marshall, C. A performance evaluation of Nextera XT and KAPA HyperPlus for rapid Illumina library preparation of long-range mitogenome amplicons. Forensic Sci. Int. Genet. 2017, 29, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Woerner, A.E.; Ambers, A.; Wendt, F.R.; King, J.L.; Moura-Neto, R.S.; Silva, R.; Budowle, B. Evaluation of the precision ID mtDNA whole genome panel on two massively parallel sequencing systems. Forensic Sci. Int. Genet. 2018, 36, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Strobl, C.; Churchill Cihlar, J.; Lagacé, R.; Wootton, S.; Roth, C.; Huber, N.; Schnaller, L.; Zimmermann, B.; Huber, G.; Lay Hong, S.; et al. Evaluation of mitogenome sequence concordance, heteroplasmy detection, and haplogrouping in a worldwide lineage study using the Precision ID mtDNA Whole Genome Panel. Forensic Sci. Int. Genet. 2018, 36, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cubero, M.J.; Saiz, M.; Martinez-Gonzalez, L.J.; Alvarez, J.C.; Eisenberg, A.J.; Budowle, B.; Lorente, J.A. Genetic Identification of Missing Persons: DNA Analysis of Human Remains and Compromised Samples. Pathobiology 2012, 79, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Brandhagen, M.D.; Just, R.S.; Irwin, J.A. Validation of NGS for mitochondrial DNA casework at the FBI Laboratory. Forensic Sci. Int. Genet. 2020, 44, 102151. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.X.; Sivakumar, I.K.A.; Maragh, S.; Veeramachaneni, V.; Hariharan, R.; Parulekar, M.; Fredrikson, K.M.; Harkins, T.T.; Lin, J.; Feldman, A.B.; et al. Next-Generation Sequencing of Human Mitochondrial Reference Genomes Uncovers High Heteroplasmy Frequency. PLoS Comput. Biol. 2012, 8, e1002737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavala, E.A.-O.; Rajagopal, S.; Perry, G.H.; Kruzic, I.; Bašić, Ž.; Parsons, T.J.; Holland, M.M. Impact of DNA degradation on massively parallel sequencing-based autosomal STR, iiSNP, and mitochondrial DNA typing systems. Int. J. Leg. Med. 2019, 133, 1369–1380. [Google Scholar] [CrossRef]
- Davis, C.; Peters, D.; Warshauer, D.; King, J.; Budowle, B. Sequencing the hypervariable regions of human mitochondrial DNA using massively parallel sequencing: Enhanced data acquisition for DNA samples encountered in forensic testing. Leg. Med. 2015, 17, 123–127. [Google Scholar] [CrossRef]
- Cihlar, J.A.-O.X.; Amory, C.A.-O.X.; Lagacé, R.; Roth, C.; Parson, W.A.-O.; Budowle, B. Developmental Validation of a MPS Workflow with a PCR-Based Short Amplicon Whole Mitochondrial Genome Panel. Genes 2020, 11, 1345. [Google Scholar] [CrossRef]
- Cihlar, J.C.; Strobl, C.; Lagacé, R.; Muenzler, M.; Parson, W.; Budowle, B. Distinguishing mitochondrial DNA and NUMT sequences amplified with the precision ID mtDNA whole genome panel. Mitochondrion 2020, 55, 122–133. [Google Scholar] [CrossRef]
- Harihara, S.; Hirai, M.; Suutou, Y.; Shimizu, K.; Omoto, K. Frequency of a 9-bp deletion in the mitochondrial DNA among Asian populations. Hum. Biol. 1992, 64, 161–166. [Google Scholar]
- Benton, M.; Macartney-Coxson, D.; Eccles, D.; Griffiths, L.; Chambers, G.; Lea, R. Complete mitochondrial genome sequencing reveals novel haplotypes in a Polynesian population. PLoS ONE 2012, 7, e35026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underhill, P.A.; Passarino, G.; Lin, A.A.; Marzuki, S.; Oefner, P.J.; Cavalli-Sforza, L.L.; Chambers, G.K. Maori origins, Y-chromosome haplotypes and implications for human history in the Pacific. Hum. Mutat. 2001, 17, 271–280. [Google Scholar] [CrossRef]
- Jäger, A.C.; Alvarez, M.L.; Davis, C.P.; Guzmán, E.; Han, Y.; Way, L.; Walichiewicz, P.; Silva, D.; Pham, N.; Caves, G.; et al. Developmental validation of the MiSeq FGx Forensic Genomics System for Targeted Next Generation Sequencing in Forensic DNA Casework and Database Laboratories. Forensic Sci. Int. Genet. 2017, 28, 52–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illumina. Illumina Sequencing Technology. 2010. Available online: https://www.illumina.com/documents/products/techspotlights/techspotlight_sequencing.pdf (accessed on 1 September 2020).
- Illumina. An introduction to Next-Generation Sequencing Technology. 2017. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/illumina_sequencing_introduction.pdf (accessed on 1 September 2020).
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.J.; Reid-Bayliss, K.S.; Emond, M.J.; Loeb, L.A. Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl. 2014, 1, 1000106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, J.; Baillie, G.; Watson, S.J.; Kellam, P.; Rambaut, A.; Robertson, D.L. Analysis of high-depth sequence data for studying viral diversity: A comparison of next generation sequencing platforms using Segminator II. BMC Bioinform. 2012, 13, 47. [Google Scholar] [CrossRef] [Green Version]
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S rRNA-Based Bacterial Community Profiling. Appl. Environ. Microbiol. 2014, 80, 7583. [Google Scholar] [CrossRef] [Green Version]
- Scientific Working Group on DNA Analysis Methods. Interpretation Guidelines for Mitochondrial DNA Analysis by Forensic DNA Testing Laboratories. 2019. Available online: https://1ecb9588-ea6f-4feb-971a-73265dbf079c.filesusr.com/ugd/4344b0_f61de6abf3b94c52b28139bff600ae98.pdf (accessed on 23 July 2019).
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, 1–12. [Google Scholar] [CrossRef] [Green Version]
- MitoMap. MITOMAP, 24 May 2019 Update. Available online: https://www.mitomap.org/MITOMAP (accessed on 24 June 2019).
- Carrasco, P.; Inostroza, C.; Didier, M.; Godoy, M.; Holt, C.L.; Tabak, J.; Loftus, A. Optimizing DNA recovery and forensic typing of degraded blood and dental remains using a specialized extraction method, comprehensive qPCR sample characterization, and massively parallel sequencing. Int. J. Leg. Med. 2020, 134, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Stray, J.; Holt, A.; Brevnov, M.; Calandro, L.M.; Furtado, M.R.; Shewale, J.G. Extraction of high quality DNA from biological materials and calcified tissues. Forensic Sci. Int. Genet. Suppl. Ser. 2009, 2, 159–160. [Google Scholar] [CrossRef]
- Loreille, O.M.; Diegoli, T.M.; Irwin, J.A.; Coble, M.D.; Parsons, T.J. High efficiency DNA extraction from bone by total demineralization. Forensic Sci. Int. Genet. 2007, 1, 191–195. [Google Scholar] [CrossRef]
- Resource, I.T.I.G.S. 1000 Genomes FTP Site. Available online: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ (accessed on 29 July 2019).
- Consortium, G.P. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, B.C.; Hancock, D.K.; Holland, K.A.; Cheng, H.; Richie, K.L. Human Mitochondrial DNA—Amplification and Sequencing Standard Reference Materials—SRM 2392 and SRM 2392-I. Nist Spec. Publ. 2003, 260, 155. [Google Scholar]
- Riman, S.; Kiesler, K.M.; Borsuk, L.A.; Vallone, P.M.; Kiesler, K.M.; Riman, S.; Borsuk, L.A. Characterization of NIST human mitochondrial DNA SRM-2392 and SRM-2392-I standard reference materials by next generation sequencing. Forensic Sci. Int. Genet. 2017, 29, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peck, M.A.; Sturk-Andreaggi, K.; Thomas, J.T.; Oliver, R.S.; Barritt-Ross, S.; Marshall, C. Developmental validation of a Nextera XT mitogenome Illumina MiSeq sequencing method for high-quality samples. Forensic Sci. Int. Genet. 2018, 34, 25–36. [Google Scholar] [CrossRef]
- Ewing, M.H.S.; Shaw, J.; Mclaren, R.S.; Storts, D.R. The PowerSeq™ CRM Nested System: A Simplified Approach to Massively Parallel Sequencing the Human mtDNA Control Region. Available online: https://promega.media/-/media/files/products-and-services/genetic-identity/ishi-29-poster-abstracts/071-ewing.pdf (accessed on 28 October 2018).
- Verogen. ForenSeq Universal Analysis Software v2.0 Reference Guide. Revision B. 2020. Available online: https://verogen.com/wp-content/uploads/2020/08/forenseq-universal-analysis-software-v2-0-reference-guide-vd2019002-b.pdf (accessed on 1 September 2020).
- Verogen. ForenSeq mtDNA Control Region Kit Reference Guide. Revision A. 2019. Available online: https://verogen.com/wp-content/uploads/2020/08/forenseq-mtdna-control-region-kit-reference-guide-vd2018024-b.pdf (accessed on 23 July 2019).
- Verogen. ForenSeq mtDNA Whole Genome Kit Reference Guide. Revision A. 2020. Available online: https://verogen.com/wp-content/uploads/2020/08/forenseq-mtdna-whole-genome-kit-reference-guide-vd2020006-a.pdf (accessed on 1 September 2020).
- Verogen. MiSeq FGx Sequencing System Reference Guide. Revision C. 2019. Available online: https://verogen.com/wp-content/uploads/2020/07/miseq-fgx-system-reference-guide-VD2018006-d.pdf (accessed on 23 July 2019).
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lascaro, D.; Castellana, S.; Gasparre, G.; Romeo, G.; Saccone, C.; Attimonelli, M. The RHNumtS compilation: Features and bioinformatics approaches to locate and quantify Human NumtS. BMC Genom. 2008, 9, 267. [Google Scholar] [CrossRef] [Green Version]
- Attimonelli, M.; Accetturo, M.; Santamaria, M.; Lascaro, D.; Scioscia, G.; Pappadà, G.; Russo, L.; Zanchetta, L.; Tommaseo-Ponzetta, M. HmtDB, a Human Mitochondrial Genomic Resource Based on Variability Studies Supporting Population Genetics and Biomedical Research. BMC Bioinform. 2005, 6, S4. [Google Scholar] [CrossRef] [Green Version]
- Clima, R.; Preste, R.; Calabrese, C.; Diroma, M.A.; Santorsola, M.; Scioscia, G.; Simone, D.; Shen, L.; Gasparre, G.; Attimonelli, M. HmtDB 2016: Data update, a better performing query system and human mitochondrial DNA haplogroup predictor. Nucleic Acids Res. 2017, 45, D698–D706. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, T.L.; Tatusov, R.L.; Zhang, J. Applications of network BLAST server. Methods Enzymol. 1996, 266, 131–141. [Google Scholar] [PubMed]
- Zhang, J.; Madden, T.L. PowerBLAST: A new network BLAST application for interactive or automated sequence analysis and annotation. Genome Res. 1997, 7, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Parson, W.; Dür, A. EMPOP—a forensic mtDNA database. Forensic Sci. Int. Genet. 2007, 1, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Gusmao, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M. DNA Commission of the International Society for Forensic Genetics: Revised and extended guidelines for mitochondrial DNA typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef]
- Wallin, J.M.; Holt, C.L.; Lazaruk, K.D.; Nguyen, T.H.; Walsh, P.S. Constructing universal multiplex PCR systems for comparative genotyping. J. Forensic Sci. 2002, 47, 52–65. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name, Source | Control Region Multiplex | mtGenome Multiplex | ||||||
|---|---|---|---|---|---|---|---|---|
| CR Coverage | CR Observed Variants | Haplogroup | mtGenome Coverage | mtGenome No Call Region(s) | mtGenome CR Observed Variants | Haplogroup | ||
| CONTEMPORARY SAMPLES | Tooth 1661, InnoGenomics | 100% | 73G 150T 152C 263G 315.1C 523c 524a 16124C 16223T 16311C 16399G | L3d1b1 | 99% | 5086–5177 | 73G 150T 152C 263G 315.1C 497M 1 523c 524a 16124C 16223T 16311C 16399G | L3d1b1 |
| Tooth 1662, InnoGenomics | 100% | 73G 153G 195C 225A 226C 263G 309.1c 315.1C 16189c 2 16193.1c 16223T 16278T 16519C | X2 + 225 | 99% | 8290–8379 | 73G 153G 195C 225A 226C 263G 309.1c 315.1C 16189c 16193.1c 16223T 16278T 16519C | X2b4a1 | |
| Tooth 1663, InnoGenomics | 100% | 73G 150T 152C 195C 198T 263G 315.1C 16189c 16223T 16320T 16519C | L3e2a1 | 100% | 73G 150T 152C 195C 198T 263G 315.1C 16189c 16223T 16320T 16519C | L3e2a1b3 | ||
| Tooth 1664, InnoGenomics | 100% | 73G 146C 152C 195C 263G 309.1C 315.1C 378Y 507C 16223T 16278T 16286T 16294T 16309G 16390A 16519C | L2a1a2 | 98% | 519, 4044–4175, 7216–7367 | 73G 146C 152C 195C 263G 309.1C 315.1C 378Y 507C 16223T 16278T 16286T 16294T 16309G 16390A 16519C | L2a1a2b | |
| Tooth 1665, InnoGenomics | 100% | 64T 93G 185A 189G 200G 236C 247A 263G 315.1C 523a 524c 16129A 16148T 16168T 16172C 16187T 16188G 16189C 16223T 16230G 16311C 16320T 16325C 16362C | L0a1a + 200 | 97% | 4044–4175, 4299–4379, 7021–7182, 7192–7196, 7206, 7216–7367 | 64T 93G 185A 189G 200G 236C 247A 263G 315.1C 523a 524c 16129A 16148T 16168T 16172C 16187T 16188G 16189C 16223T 16230G 16311C 16320T 16325C 16362C | L0a1a2 | |
| Bone S1, Commercially cremated, SHSU | 100% | 73G 150T 263G 315.1C 16189c 16193.1c 16270T 16398A | U5b2a2 | 99% | 5307, 5327, 5334–5338, 5343, 6718−6810, 7308−7310, 12,563−12,564, 15,571−15,573 | 73G 150T 263G 315.1C 16183M 3 16189c 16193.1c 16270T 16398A | U5b2a2b | |
| Bone S2, embalmed, SHSU | 100% | 73G 150T 185A 228A 263G 295T 309.1C 315.1C 462T 489C 16069T 16126C | J1c | 97% | 1103, 1132−1138, 1150, 1164, 1172, 2661−2663, 3606, 5307−5347, 6121, 6139, 6718−6810, 7256−7342, 7508−7559, 11,187−11,189, 12,466−12,614, 15,190, 15,539−15,581 | 73G 150T 185A 228A 263G 295T 309.1C 315.1C 462T 489C 16069T 16126C | J1c | |
| Bone S3, embalmed, SHSU | 100% | 73G 143A 146C 152C 189G 195C 263G 315.1C 16129A 16189c 16192T 16223T 16278T 16294T 16309G 16390A | L2a1 | 98% | 4044−4175, 5081−5177, 5335−5336, 7216−7310 | 73G 143A 146C 152C 189G 195C 263G 315.1C 16129A 16189c 16192T 16223T 16278T 16294T 16309G 16390A | L2a1n | |
| Bone S4, SHSU | 100% | 73G 152C 263G 315.1C 16093Y 16256T 16270T 16399G | U5a1 | 100% | 73G 152C 263G 315.1C 16093Y 16256T 16270T 16399G | U5a1a1b | ||
| Bone S5, burned, SHSU | 100% | 195C 263G 315.1C 523a 524c | R0 | 99% | 5858−5975, 8444−8446, 12,466−12536, 12,563−12,614 | 195C 263G 315.1C 523a 524c | H4a1a4b | |
| Bone S6, burned, SHSU | 100% | 73G 263G 309.1c 315.1C 16126C 16294T 16296T 16519C | T2 | 100% | 2663, 3550−3606, 5334−5337, 7308−7310, 15,571−15,574 | 73G 263G 309.1c 315.1C 481Y 4 16126C 16294T 16296T 16519C | T2a1a | |
| Bone S7, burned, SHSU | 100% | 263G 309.1c 315.1C 316A 16291T 16519C | H1j2a | 100% | 15,572 | 263G 309.1c 309.2c 5 315.1C 316A 16291T 16519C | H1j2a | |
| ANCIENT SAMPLES | Interred bone P2, PSU | 100% | 73G 6 263G 315.1c 7 489Y 8 16192Y 16256Y 16260Y 16270T 16291T 16399R | U5a1b1 | 96% | 1094−1177, 2668−2671, 3590−3591, 5307−5346, 6109−6141, 6719−6810, 7256−7342, 7545, 8291−8379, 11,193−11197, 12,466−12614, 15,519−15,581 | 73R 6 263G 315.1C 7 523a 9 16076M 10 16192Y 16256Y 16260Y 16270T 16291T 16399R | U5a1b1c |
| Interred bone P43pt1, PSU | 100% | 152C 263G 309.1c 315.1c 16234T 16270Y | H | 99% | 5340−5344, 6718−6810, 7314−7318, 15,579 | 152C 263G 309.1c 315.1c 495Y 11 506Y 12 16234T 16270Y | H13a1d | |
| Interred bone P48, PSU | 100% | 257R 263G 315.1C 477C 13 16093Y 16192Y 16270Y 16519C | H1c | 99% | 5307−5347, 6718−6810, 7258−7266, 7273, 7288−7340, 8345−8349, 12,555−12,559 | 257R 263G 315.1C 477Y 13 514Y 14 16093Y 16192Y 16270Y 16519C | H1 | |
| Interred bone P73, PSU | 100% | 73G 153G 195C 263G 309.1C 15 309.2c 315.1C 17 489G 16189c 16223T 16278T 16294T 16519C | X1′2′3 | 98% | 2668−2671, 5307−5347, 6109−6141, 6718−6810, 7256−7348, 12,555−12559, 15,520−15,581 | 73G 153G 195C 263G 309.1c 15 309.2c 310Y 16 315.1c 17 459Y 18 489G 494Y 496Y 497Y 511Y 513R 514Y 518Y 557Y 19 16188c 20 16189c 16223T 16278T 16294T 16519C | X2 | |
| Control Region | mtGenome | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Input | CR Coverage | CR No Call Region(s) | CR Observed Variants | Haplogroup | Concordance (Relative to Buccal) | mtG Coverage | mtGenome No Call Region(s) | mtG CR Observed Variants | Haplogroup | Concordance (Relative to Buccal) |
| Buccal sample 2 | 100 pg | 100% | 73G 146C 150T 263G 309.1c 315.1C 523a 524c 16126C 16292T 16294T 16296T 16519C | T2c1 + 146 | 100% | 73G 146C 150T 263G 309.1c 315.1C 523a 524c 16126C 16292T 16294T 16296T 16519C | T2c1e | ||||
| 0.5 cm Hair sample 2 | 12 µL | 99.9% | 310 | 73G 146C 150T 263G 315.1C 523a 524c 16126C 16292T 16294T 16296T 16519C | T2c1 + 146 | 100% | 100% | 73G 146C 150T 263G 309.1c 1 315.1C 523a 524c 16126C 16292T 16294T 16296T 16519C | T2c1e | 100% | |
| 2 cm Hair sample 2 | 12 µL | 100% | 73G 146C 150T 263G 315.1C 523a 524c 16126C 16292T 16294T 16296T 16519C | T2c1 + 146 | 100% | 100% | 73G 146C 150T 263G 309.1c 1 315.1C 523a 524c 16126C 16292T 16294T 16296T 16519C | T2c1e | 100% | ||
| Buccal sample 4 | 100pg | 99.9% | 310 | 146C 263G 309.1C 315.1C 16142T 16325C | HV | 99.7% | 9538–9590 | 146C 263G 309.1c 2 309.2c 3 315.1C 16142T 16325C | H47 | ||
| 0.5 cm Hair sample 4 | 12 µL | 96.9% | 303–346 | 146C 263G 16142T 16325C | HV | 100% | 98.7% | 8290–8379, 9538–9590, 12,496–12,601 | 146C 263G 309.1c 2 315.1C 16142T 16325C | H47 | 100% |
| 2 cm Hair sample 4 | 12 µL | 96.3% | 303–347 | 146C 263G 16142T 16325C | HV | 100% | 99.0% | 9541, 9545–9547, 9549–9550, 9552, 9555–9557, 9564, 9568, 9570–9571, 9577, 9581, 9588–9589, 12,466–12,614 | 146C 263G 309.1c 2 315.1C 16142T 16325C | H47 | 100% |
| Buccal sample 5 | 100pg | 100% | 263G 315.1C | R0 | 100% | 263G 315.1C | H4a1a1 | ||||
| 0.5 cm Hair sample 5 | 12 µL | 100% | 263G 315.1C | R0 | 100% | 100% | 263G 315.1C | H4a1a1 | 100% | ||
| 2 cm Hair sample 5 | 12 µL | 100% | 263G 315.1C | R0 | 100% | 100% | 263G 315.1C | H4a1a1 | 100% | ||
| Buccal sample 8 | 100pg | 100% | 73G 150T 194T 263G 315.1C 489C 523a 524c 16223T 16362C 16519C | D4b2b2a | 100% | 73G 150T 194T 263G 315.1C 489C 523a 524c 16223T 16362C 16519C | D4b2b2a | ||||
| 0.5 cm Hair sample 8 | 12 µL | 100% | 73G 150T 194T 263G 315.1C 489C 523a 524c 16223T 16362C 16519C | D4b2b2a | 100% | 100% | 73G 150T 194T 263G 315.1C 489C 523a 524c 16223T 16362C 16519C | D4b2b2a | 100% | ||
| 2 cm Hair sample 8 | 12 µL | 100% | 73G 150T 194T 263G 315.1C 489C 523a 524c 16223T 16362C 16519C | D4b2b2a | 100% | 100% | 73G 150T 194T 263G 315.1C 489C 523a 524c 16223T 16362C 16519C | D4b2b2a | 100% | ||
| Buccal sample 11 | 100pg | 100% | 73G 152C 249del 263G 309.1c 315.1C 523a 524c 16108T 16129A 16162G 16172C 16232A 16304C 16357C 16519C | F1a1a | 100% | 73G 152C 249del 263G 309.1c 315.1C 523a 524c 16108T 16129A 16162G 16172C 16232A 16304C 16357C 16519C | F1a1a | ||||
| 0.5 cm Hair sample 11 | 12 µL | 96.1% | 303–347 | 73G 152C 249del 263G 523a 524c 16108T 16129A 16162G 16172C 16232A 16304C 16357C 16519C | F1a1a | 100% | 99.8% | 9489–9526 | 73G 152C 249del 263G 309.1c 315.1C 523a 524c 16108T 16129A 16162G 16172C 16232A 16304C 16357C 16519C | F1a1a | 100% |
| 2 cm Hair sample 11 | 12 µL | 99.9% | 310 | 73G 152C 249del 263G 309.1c 315.1C 523a 524c 16108T 16129A 16162G 16172C 16232A 16304C 16357C 16519C | F1a1a | 100% | 100% | 73G 152C 249del 263G 309.1c 315.1C 523a 524c 16108T 16129A 16162G 16172C 16232A 16304C 16357C 16519C | F1a1a | 100% | |
| Buccal sample 12 | 100pg | 100% | 195Y 263G 309.1c 315.1C 4 16519C | R0 | 100% | 195Y 263G 309.1c 315.1C 4 16519C | H40b | ||||
| 0.5 cm Hair sample 12 | 12 µL | 96.1% | 303–347 | 195Y 263G 16519C | R0 | 100% | 98.5% | 5307, 5311–5312, 5318, 5321, 5323–5332, 6718–6810, 7256–7342, 15,519–15,581 | 195Y 263G 309.1c 315.1c 4 489Y 5 16519C | H40b | 100% |
| 2 cm Hair sample 12 | 12 µL | 100% | 195Y 263G 309.1c 315.1C 4 16519C | R0 | 100% | 99.5% | 6718–6810 | 195Y 263G 309.1c 315.1c 4 16519C | H40b | 100% | |
| ForenSeq Multiplex | gDNA Input | AT1 | Mixture Ratio | Expected Variant Allele Ratio (%) | Expected Minor Variant Range (%) | Expected Major Variant Range (%) | Expected Minor Variants (2800 M) | Observed Minor Variants | Minor Variant Detection Rate |
|---|---|---|---|---|---|---|---|---|---|
| Control region | 100 pg | 3.7% | 1:3 | 25:75 | 22–36 | 64–78 | 10 | 10 | 100% |
| 100 pg | 3.7% | 1:5 | 17:83 | 10–17 | 82–90 | 10 | 10 | 100% | |
| 100 pg | 3.7% | 1:15 | 6:94 | 4–7 | 93–96 | 10 | 10 | 100% | |
| 5 pg | 3.7% | 1:3 | 25:75 | 24–36 | 64–76 | 10 | 10 | 100% | |
| 5 pg | 3.7% | 1:5 | 17:83 | 4–19 | 81–96 | 10 | 112 | 100% | |
| 5 pg | 3.7% | 1:15 | 6:94 | 3–7 | 93–97 | 10 | 8 | 80% | |
| mtGenome | 100 pg | 6% | 1:1 | 50:50 | 26–47 | 53–74 | 27 | 27 | 100% |
| 100 pg | 6% | 1:3 | 25:75 | 11–25 | 75–89 | 27 | 27 | 100% | |
| 100 pg | 6% | 1:9 | 10:90 | 0–11 | 89–100 | 27 | 15 | 55.6% | |
| 5 pg | 6% | 1:1 | 50:50 | 24–47 | 53–76 | 27 | 27 | 100% | |
| 5 pg | 6% | 1:3 | 25:75 | 8–19 | 81–92 | 27 | 27 | 100% | |
| 5 pg | 6% | 1:9 | 10:90 | 0–8 | 92–100 | 27 | 7 | 25.9% |
| Input | Repeatability | Reproducibility | |||
|---|---|---|---|---|---|
| Control Region Multiplex | mtGenome Multiplex | Control Region Multiplex | mtGenome Multiplex | ||
| Precision | 2 pg | 100% | 97.9% 1 | 100% | 97.9% 2 |
| 20 pg | N/A | 99.4% 3 | 100% | N/A | |
| 100 pg | 100% | 99.98% 4 | 100% | 100% | |
| Concordance | 2 pg | 100% | 100% 5 | 100% | 100% 6 |
| 20 pg | N/A | 100% 7 | 100.0% | N/A | |
| 100 pg | 100% | 100% 8 | 100.0% | 100% 9 | |
| Average reads per amplicon | 2 pg | 7129 | 3580 | 6983 | 1260 |
| 20 pg | N/A | 1764 | 20,728 | N/A | |
| 100 pg | 43,401 | 3146 | 29,574 | 3645 | |
| Sample (100 pg) | Expected CR Variants [52,53,54] | Control Region Kit | Whole Genome Kit |
|---|---|---|---|
| Observed CR Variants (100% Concordance) | Observed CR Variant (100% Concordance) | ||
| CHR | 64Y 73G 195C 204C 207A 263G 309.1C 315.1C 16183C 16189C 16193.1c 16193.2c 16223T 16278T 16519C | 64Y 73G 195C 204C 207A 263G 309.1C 315.1C 16183C 16189C 16193.1c 16193.2c 16223T 16278T 16519C | 64Y 73G 195C 204C 207A 263G 309.1C 315.1C 16183C 16189C 16193.1c 16193.2c 16223T 16278T 16519C |
| 9947A | 93G 195C 214G 263G 309.1C 309.2C 315.1C 16311C 16519C | 93G 195C 214G 263G 309.1C 309.2C 315.1C 16311C 16519C | 93G 195C 214G 263G 309.1C 309.2C 315.1C 16311C 16519C |
| HL-60 | 73G 150T 152C 263G 295T 315.1C 489C 16069T 16193T 16278T 16362C | 73G 150T 152C 263G 295T 315.1C 489C 16069T 16193T 16278T 16362C | 73G 150T 152C 263G 295T 315.1C 489C 16069T 16193T 16278T 16362C |
| GM03798 | 263G 315.1C 16357C 16519C | 263G 315.1C 16357C 16519C | 263G 315.1C 16357C 16519C |
| GM10472A | 73G 185A 228A 263G 295T 315.1C 462T 482C 489C 16069T 16126C 16292T | 73G 185A 228A 263G 295T 315.1C 462T 482C 489C 16069T 16126C 16292T | 73G 185A 228A 263G 295T 315.1C 462T 482C 489C 16069T 16126C 16292T |
| Sample | Control Region Multiplex: Observed Variants | Haplogroup Based on Control Region | mtGenome Multiplex: No Call Region(s) | mtGenome Multiplex: Control Region Observed Variants | Haplogroup Based on mtGenome | mtGenome Concordance (Compared to 1KPG) | CR Concordance (Compared to CR Multiplex) |
|---|---|---|---|---|---|---|---|
| HG00181 | 73G 195C 263G 309.1C 315.1C 499A 524.1a 524.2c 16356C 16519C | U4 | 6922–6988 | 73G 195C 263G 309.1C 315.1C 499A 524.1a 524.2c 16356C 16519C | U4d1a1 | 100% | 100% |
| HG00383 | 263G 315.1C 523a 524c 16093C 16129A 16316G 16519C | H27 | 263G 315.1C 523a 524c 16093C 16129A 16316G 16519C | H27a | 100% | 100% | |
| HG00384 | 73G 150T 263G 309.1c 309.2c 315.1C 16144C 16183M 16189C 16193.1c 16193.2c 16270T | U5b1b1a | 73G 150T 263G 309.1c 309.2c 315.1C 16144C 16183M 16189C 16193.1c 16193.2c 16270T | U5b1b1a | 100% | 100% | |
| HG00844 | 73G 249del 263G 309.1C 310Y 1 315.1C 489C 16092C 16189C 16193.1c 16193.2c 16223T 16298C 16327T 16355T 16519C | C | 470–519, 3550–3606, 13,013–13,080, 15,539–15,581 | 73G 249del 263G 309.1c 310Y 315.1C 16092C 16189c 2 16193.1c 16223T 16298C 16327T 16355T 16519C | C7a | 100% | 100% |
| HG01197 | 73G 150T 263G 279C 315.1C 455.1T 517T 16224C 16270T | U5b2b3a | 73G 150T 263G 279C 315.1C 455.1T 517T 523a 16181R 16224C 16270T | U5b2b3a | 100% | 100% | |
| HG01204 | 73G 249del 290del 291del 315.1C 489C 493G 523a 524c 16223T 16298C 16325C 16327T 16519C | C1b2 | 73G 249del 290del 291del 315.1C 489C 493G 523a 524c 16223T 16298C 16325C 16327T 16519C | C1b2 | 100% | 100% | |
| HG01205 | 73G 263G 315.1C 523a 524c 16093C 16223T 16278T 16362C 16519C | L3b1a | 73G 189R 263G 315.1C 523a 524c 16093C 16223T 16278T 16362C 16519C | L3b1a + @16124 | 100% | 100% | |
| HG01497 | 73G 263G 309.1C 309.2c 315.1c 498del 499A 524.1a 524.2c 16183c 16189C 16193.1c 16217C 16519C | B2d | 73G 263G 309.1C 309.2c 315.1c 498del 499A 524.1a 524.2c 16183c 16189C 16193.1c 16217C 16519C | B2d | 100% | 100% | |
| HG01498 | 73G 263G 307c 308c 309c 310c 498del 499A 16182c 16183c 16189C 16193.1c 16217C 16519C | B2d | 73G 263G 307c 308c 309c 310c 498del 499A 16182c 16183c 16189C 16193.1c 16217C 16519C | B2d | 100% | 100% | |
| HG01550 | 73G 263G 309.1C 309.2c 309.3c 310Y 315.1C 498del 499A 16182C 16183c 16189C 16193.1c 16217C 16519C | B2d | 73G 263G 309.1C 309.2c 309.3c 310Y 315.1C 498del 499A 16182c 16183c 16189C 16193.1c 16217C 16519C | B2d | 100% | 100% | |
| HG01551 | 73G 150T 263G 315.1C 523a 524c 16051G 16223T 16264T 16519C | L3e4a | 73G 150T 263G 315.1C 523a 524c 16051G 16223T 16264T 16519C | L3e4a | 100% | 100% | |
| HG01790 | 263G 309.1C 309.2C 315.1C | R0 | 263G 309.1C 309.2c 315.1C | H33a | 100% | 100% | |
| HG02190 | 73G 150T 263G 315.1C 489C 523a 524c 16172C 16182C 16183c 16189C 16193.1c 16223T 16362C 16519C | D5a2 | 73G 150T 263G 315.1C 489C 523a 524c 16172C 16182c 16183c 16189C 16193.1c 16223T 16362C 16519C | D5a2b | 100% | 100% | |
| HG02215 | 263G 315.1C 16311C 16519C | R0 | 263G 315.1C 16311C 16519C | H3m | 100% | 100% | |
| HG02236 | 214G 263G 315.1C 16172C 16519C | HV | 214G 263G 315.1C 16172C 16519C | H1 | 100% | 100% | |
| HG02238 | 263G 309.1C 309.2C 315.1C 16129A 16519C | H | 263G 309.1C 309.2c 315.1C 16129A 16519C | H1j1 | 100% | 100% | |
| HG02239 | 263G 292C 309.1C 315.1C 16519C | R0 | 263G 292C 309.1c 315.1C 16519C | H1 | 100% | 100% | |
| HG02317 | 73G 143A 146C 152C 195C 263G 309.1C 315.1C 16129A 16223T 16278T 16294T 16309G 16390A | L2a1c + 16129 | 73G 143A 146C 152C 195C 263G 309.1C 315.1C 16129A 16223T 16278T 16294T 16309G 16390A | L2a1c5 | 100% | 100% | |
| HG02322 | 73G 89C 93G 95C 152C 182T 186A 189C 236C 247A 263G 297G 315.1C 316A 523a 524c 16129A 16182C 16183c 16189C 16223T 16235G 16274A 16278T 16293G 16294T 16311C 16360T 16519C | L1c1a2 | 4044–4175, 7256–7367 | 73G 89C 93G 95C 152C 182T 186A 189C 236C 247A 263G 297G 315.1C 316A 523a 524c 16129A 16182c 16183c 16189C 16223T 16235G 16274A 16278T 16293G 16294T 16311C 16360T 16519C | L1c1a2 | 100% | 100% |
| HG02449 | 73G 150T 263G 273Y 309.1C 315.1C 523a 524c 16051G 16223T 16264T 16519C | L3e4a | 73G 150T 263G 273Y 309.1C 315.1C 523a 524c 16051G 16223T 16264T 16519C | L3e4a | 100% | 100% | |
| HG02450 | 73G 150T 195C 263G 309.1C 315.1C 499A 16223T 16320T 16399G 16519C | L3e2a1b1 | 73G 150T 195C 263G 309.1C 315.1C 499A 16223T 16320T 16399G 16519C | L3e2a1b1 | 100% | 100% | |
| HG02513 | 73G 249del 263G 309.1C 315.1C 521a 522c 523a 524c 16172C 16304C 16465T 16519C | F1a2a | 73G 249del 263G 309.1c 315.1C 521a 522c 523a 524c 16172C 16304C 16465T 16519C | F1a2a | 100% | 100% | |
| HG02521 | 73G 150T 263G 309.1c 315.1C 16111T 16129A 16223T 16257A 16261T | N9a1 | 73G 150T 263G 309.1c 315.1C 16111T 16129A 16223T 16257A 16261T | N9a1 | 100% | 100% | |
| HG03369 | 73G 150T 195C 263G 315.1C 16223T 16265T 16519C | L3e3 | 73G 150T 195C 263G 315.1C 16223T 16265T 16519C | L3e3b | 100% | 100% | |
| HG03370 | 73G 263G 315.1C 372C 523a 524c 16124C 16223T 16278T 16519C | L3 | 73G 263G 315.1C 372C 523a 524c 16124C 16223T 16278T 16519C | L3b1a | 100% | 100% | |
| HG03372 | 73G 150T 195C 263G 315.1C 16223T 16265T 16519C | L3e3 | 73G 150T 195C 263G 315.1C 16223T 16265T 16519C | L3e3b | 100% | 100% | |
| HG03577 | 73G 150T 195C 263G 309.1C 315.1C 16177G 16223T 16311C 16320T 16354T 16519C | L3e2 | 73G 150T 195C 263G 309.1C 315.1C 16177G 16223T 16311C 16320T 16354T 16519C | L3e2a | 100% | 100% | |
| HG03578 | 73G 146C 152C 195C 263G 315.1C 524.1a 524.2c 524.3a 524.4c 16223T 16233G 16278T 16294T 16309G 16368C 16390A 16519C | L2a1a1 | 73G 146C 152C 195C 263G 315.1C 524.1a 524.2c 524.3a 524.4c 16223T 16233G 16278T 16294T 16309G 16368C 16390A 16519C | L2a1a1 | 100% | 100% | |
| HG03583 | 73G 189C 195C 263G 315.1C 523del 524c 16126C 16179T 16215G 16223T 16256A 16284G 16311C | L3h1b1a | 73G 189C 195C 263G 315.1C 523del 524c 16126C 16179T 16215G 16223T 16256A 16284G 16311C | L3h1b1a | 100% | 100% | |
| HG03594 | 16T 73G 93G 188G 200G 204C 263G 309.1C 315.1C 489C 16153A 16223T 16287T 16327A 16519C | M91a | 16T 73G 93G 188G 200G 204C 263G 309.1C 315.1C 489C 16153A 16223T 16287T 16327A 16519C | M91a | 100% | 100% | |
| HG03595 | 41T 73G 153G 263G 309.1C 315.1C 489C 16223T 16234T 16295G 16311C 16320T 16519C | M | 41T 73G 153G 263G 309.1C 315.1C 489C 16223T 16234T 16295G 16311C 16320T 16519C | M49 | 100% | 100% | |
| HG03600 | 73G 195A 263G 315.1C 489C 523a 524c 16179del 16223T 16519C | M30 | 73G 195A 263G 315.1C 489C 523a 524c 16179del 16223T 16519C | M30d1 | 100% | 100% | |
| NA12812 | 44.1C 263G 309.1C 309.2C 315.1C 16093C 16129A 16183C 16189C 16193.1c 16519C | HV | 44.1C 263G 309.1C 309.2C 315.1C 16093C 16129A 16183C 16189C 16193.1c 16519C | H1 + 16189 | 100% | 100% | |
| NA12813 | 73G 263G 309.1C 315.1C 16114A 16129A 16189c 16192Y 16192.1t 16256T 16270T 16294T 16526A | U5a2a | 73G 263G 309.1C 315.1C 16114A 16129A 16189c 16192Y 16192.1t 16256T 16270T 16294T 16526A | U5a2a | 100% | 100% | |
| NA12814 | 73G 263G 315.1C 16192T 16256T 16270T 16291T 16399G | U5a1b1 | 73G 263G 315.1C 16192T 16256T 16270T 16291T 16399G | U5a1b1a2 | 100% | 100% | |
| NA12815 | 73G 263G 315.1C 16129A 16316G 16519C | H | 73G 263G 315.1C 16129A 16316G 16519C | H27 | 100% | 100% | |
| NA12872 | 263G 309.1C 309.2c 315.1C 16172C 16311C | HV | 263G 309.1C 309.2c 315.1C 16172C 16311C | HV6 | 100% | 100% | |
| NA12873 | 152C 195C 263G 309.1c 309.2c 315.1C 16293G 16311C 16525G | H11a6 | 152C 195C 263G 309.1c 309.2c 315.1C 16293G 16311C 16525G | H11a6 | 100% | 100% | |
| NA12874 | 73G 185A 188G 228A 263G 295T 309.1C 315.1C 462T 489C 16069T 16126C 16319A | J1c | 73G 185A 188G 228A 263G 295T 309.1C 315.1C 462T 489C 523a 16069T 16126C 16319A | J1c8a | 100% | 100% | |
| NA19240 | 73G 150T 152C 195C 263G 315.1C 16172C 16183c 16189C 16193.1c 16223T 16293T 16320T 16519C | L3e2b | 73G 150T 152C 195C 263G 315.1C 16172C 16183c 16189C 16193.1c 16223T 16293T 16320T 16519C | L3e2b5 | 100% | 100% | |
| NA20346 | 73G 150T 195C 263G 315.1C 16145A 16172C 16189c 16193.1c 16193.2c 16223T 16320T 16519C | L3e2b | 73G 150T 195C 263G 315.1C 16145A 16172C 16189c 16193.1c 16193.2c 16223T 16320T 16519C | L3e2b1a1 | 100% | 100% | |
| NA20356 | 73G 263G 309.1c 315.1C 16172C 16219G 16278T 16291Y 16519C | U6a | 73G 263G 309.1c 315.1C 16172C 16219G 16278T 16291Y 16519C | U6a5 | 100% | 100% | |
| NA20509 | 263G 309.1C 309.2C 309.3c 310Y 315.1C 523a 524c 16182C 16183c 16189C 16193.1c 16261T 16274A 16356C 16519C | H1b | 263G 309.1C 309.2C 309.3c 315.1C 523a 524c 16182c 16183c 16189C 16193.1c 16193.2c 16261T 16274A 16356C 16519C | H1b1 | 100% | 100% | |
| NA20510 | 73G 189G 195C 204C 207A 263G 315.1C 16192T 16223T 16309G 16325C 16519C | W6 | 73G 189G 195C 204C 207A 263G 315.1C 16192T 16223T 16309G 16325C 16519C | W6 | 100% | 100% | |
| NA20828 | 73G 263G 309.1c 315.1C 497T 524.1a 524.2c 524.3a 524.4c 16129A 16177G 16224C 16311C 16390A 16519C | K1a12a1a | 73G 263G 309.1c 315.1C 497T 524.1a 524.2c 524.3a 524.4c 16129A 16177G 16224C 16311C 16390A 16519C | K1a4f1 | 100% | 100% | |
| NA20832 | 146C 263G 309.1C 309.2c 315.1C 16519C | HV | 146C 263G 309.1C 309.2c 315.1C 16519C | H1n | 100% | 100% | |
| NA20845 | 73G 152C 263G 309.1c 315.1C 489C 16086C 16129A 16223T 16519C | M | 73G 152C 263G 309.1c 315.1C 489C 16086C 16129A 16223T 16519C | M5a2a | 100% | 100% | |
| NA21143 | 73G 146C 263G 309.1C 309.2c 315.1C 489C 16129A 16223T 16320T | M | 73G 146C 263G 309.1C 309.2c 315.1C 489C 16129A 16223T 16320T | M5c1 | 100% | 100% | |
| NA21144 | 73G 195C 263G 315.1C 16093C 16519C | R8 | 73G 195C 263G 315.1C 16093C 16519C | R8a1b | 100% | 100% |
| L Lineages “African” | M Lineages “Asian” | N Lineages “Eurasian” | ||||||
|---|---|---|---|---|---|---|---|---|
| hg | # | % | hg | # | % | hg | # | % |
| L3 | 2135 | 35.6% | M | 5250 | 50% | H | 9167 | 28% |
| L0 | 1500 | 25% | D | 2358 | 22% | U | 4231 | 13% |
| L2 | 1322 | 22% | C | 1651 | 16% | B | 4193 | 13% |
| L1 | 878 | 14.7% | E | 456 | 4% | J | 2319 | 7% |
| L4 | 105 | 1.8% | G | 437 | 4% | T | 2237 | 7% |
| L5 | 39 | 0.7% | Z | 191 | 2% | K | 1817 | 6% |
| L6 | 12 | 0.2% | Q | 177 | 2% | F | 1663 | 5% |
| Total | 5991 | 100% | Total | 10,520 | 100% | A | 1386 | 4% |
| Overall 12% (5991/48,882) | Overall 22% (10,520/48,882) | R | 1077 | 3% | ||||
| N | 785 | 2% | ||||||
| HV | 735 | 2% | ||||||
| I | 718 | 2% | ||||||
| V | 693 | 2% | ||||||
| W | 529 | 2% | ||||||
| X | 470 | 1% | ||||||
| P | 159 | 0.5% | ||||||
| Y | 135 | 0.4% | ||||||
| S | 49 | 0.2% | ||||||
| O | 8 | 0.02% | ||||||
| Total | 32,371 | 100% | ||||||
| Overall 66% (32,371/48,882) | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holt, C.L.; Stephens, K.M.; Walichiewicz, P.; Fleming, K.D.; Forouzmand, E.; Wu, S.-F. Human Mitochondrial Control Region and mtGenome: Design and Forensic Validation of NGS Multiplexes, Sequencing and Analytical Software. Genes 2021, 12, 599. https://doi.org/10.3390/genes12040599
Holt CL, Stephens KM, Walichiewicz P, Fleming KD, Forouzmand E, Wu S-F. Human Mitochondrial Control Region and mtGenome: Design and Forensic Validation of NGS Multiplexes, Sequencing and Analytical Software. Genes. 2021; 12(4):599. https://doi.org/10.3390/genes12040599
Chicago/Turabian StyleHolt, Cydne L., Kathryn M. Stephens, Paulina Walichiewicz, Keenan D. Fleming, Elmira Forouzmand, and Shan-Fu Wu. 2021. "Human Mitochondrial Control Region and mtGenome: Design and Forensic Validation of NGS Multiplexes, Sequencing and Analytical Software" Genes 12, no. 4: 599. https://doi.org/10.3390/genes12040599
APA StyleHolt, C. L., Stephens, K. M., Walichiewicz, P., Fleming, K. D., Forouzmand, E., & Wu, S.-F. (2021). Human Mitochondrial Control Region and mtGenome: Design and Forensic Validation of NGS Multiplexes, Sequencing and Analytical Software. Genes, 12(4), 599. https://doi.org/10.3390/genes12040599