
The Role of Wild-Type RAS in Oncogenic RAS Transformation


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
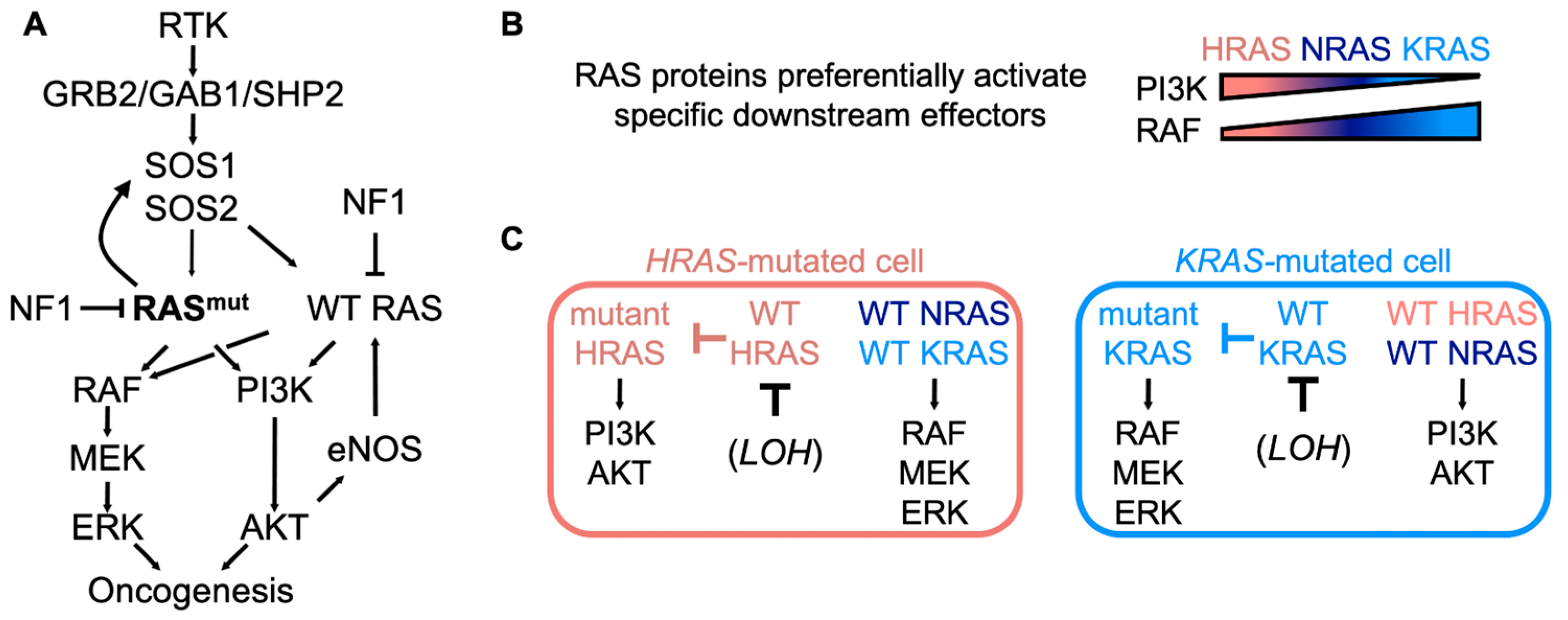
2. Contributions of WT RAS to Mutant RAS-Driven Cancers
2.1. The WT RAS Allele of the Same Isoform as Mutated RAS Inhibits Tumorigenesis
2.2. WT RAS Family Members Distinct from the Mutated RAS Allele Promote Oncogenesis
3. Mechanisms of WT RAS Activation in RAS-Mutated Cancers
3.1. Mutant RAS-Dependent and RTK-Dependent Mechanisms Activate WT RAS in RAS-Mutated Tumor Cells
3.2. The RasGEFs SOS1 and SOS2 May Play Non-Overlapping Roles in Cells Expressing Oncogenic RAS
3.3. WT RAS Cooperates with Mutant RAS to Fully Activate Downstream RAS Effector Pathways
4. WT RAS Signaling Underlies Resistance to Targeted Therapies in RAS-Mutated Cancers
4.1. Inhibitors of RAS Effector Pathways
4.2. Mutant RAS Inhibition
4.2.1. Tipifarnib as an HRAS-Specific Inhibitor
4.2.2. Covalent KRASG12C Inhibitors
4.3. Inhibition of Proximal RTK Signaling Can Overcome MEK- and KRASG12C-Inhibitor Resistance
4.4. SOS1 and SHP2 Are Therapeutic Targets in RAS-Mutated Cancer Cells
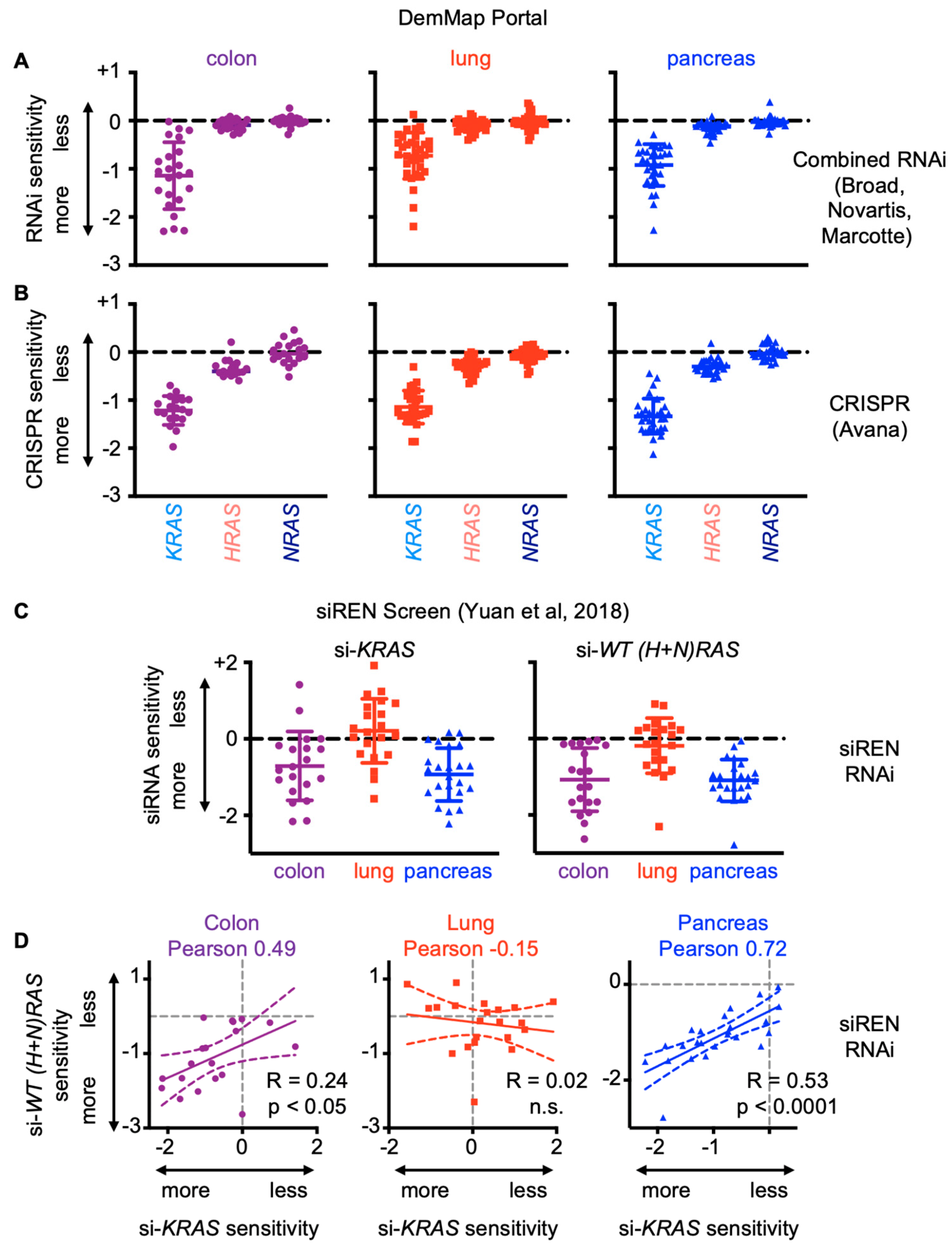
4.5. The Spectrum of KRAS Mutations between Different Cancer Types Leads to Cancer-Specific Vulnerabilities to WT RAS Inhibition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor-tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papke, B.; Der, C.J. Drugging ras: Know the enemy. Science 2017, 355, 1158–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The frequency of ras mutations in cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Riely, G.J.; Marks, J.; Pao, W. Kras mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Markowitz, S.D. Genetic and epigenetic alterations in colon cancer. Annu. Rev. Rev. Genom. Hum. Genet. 2002, 3, 101–128. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Hruban, R.H.; Canto, M.; Kern, S.E. Focus on pancreas cancer. Cancer Cell 2002, 2, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA: A Cancer Journal for Clinicians. Cancer Stat. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The origins and genetic interactions of kras mutations are allele- and tissue-specific. Nat. Commun. 2021, 12, 1808. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. Ras oncogenes: Weaving a tumorigenic web. Nat. Rev. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Knickelbein, K.; Zhang, L. Mutant kras as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015, 2, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, K.M. Kras alleles: The devil is in the detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.E.; Gandhi, L.; Costa, D.B. Management and future directions in non-small cell lung cancer with known activating mutations. Am. Soc. Clin. Oncol. Educ. Book 2014, e353-65. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable ras: Mission possible? Nat. Rev. Rev. Drug. Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-ras(g12c) inhibitors allosterically control gtp affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting kras mutant cancers with a covalent g12c-specific inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical kras(g12c) inhibitor amg 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The kras(g12c) inhibitor mrtx849 provides insight toward therapeutic susceptibility of kras-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- McCormick, F. Progress in targeting ras with small molecule drugs. Biochem. J. 2019, 476, 365–374. [Google Scholar] [CrossRef]
- Orgovan, Z.; Keseru, G.M. Small molecule inhibitors of ras proteins with oncogenic mutations. Cancer Metastasis Rev. 2020, 39, 1107–1126. [Google Scholar] [CrossRef]
- Zhou, B.; Der, C.J.; Cox, A.D. The role of wild type ras isoforms in cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Spandidos, D.A.; Frame, M.; Wilkie, N.M. Expression of the normal h-ras1 gene can suppress the transformed and tumorigenic phenotypes induced by mutant ras genes. Anticancer Res. 1990, 10, 1543–1554. [Google Scholar]
- To, M.D.; Rosario, R.D.; Westcott, P.M.; Banta, K.L.; Balmain, A. Interactions between wild-type and mutant ras genes in lung and skin carcinogenesis. Oncogene 2013, 32, 4028–4033. [Google Scholar] [CrossRef] [Green Version]
- Diaz, R.; Ahn, D.; Lopez-Barcons, L.; Malumbres, M.; de Castro, I.P.; Lue, J.; Ferrer-Miralles, N.; Mangues, R.; Tsong, J.; Garcia, R.; et al. The n-ras proto-oncogene can suppress the malignant phenotype in the presence or absence of its oncogene. Cancer Res. 2002, 62, 4514–4518. [Google Scholar]
- Wang, J.; Liu, Y.; Li, Z.; Wang, Z.; Tan, L.X.; Ryu, M.J.; Meline, B.; Du, J.; Young, K.H.; Ranheim, E.; et al. Endogenous oncogenic nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood 2011, 118, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Diaz, R.; Lue, J.; Mathews, J.; Yoon, A.; Ahn, D.; Garcia-Espana, A.; Leonardi, P.; Vargas, M.P.; Pellicer, A. Inhibition of ras oncogenic activity by ras protooncogenes. Int. J. Cancer 2005, 113, 241–248. [Google Scholar] [CrossRef]
- To, M.D.; Perez-Losada, J.; Mao, J.H.; Hsu, J.; Jacks, T.; Balmain, A. A functional switch from lung cancer resistance to susceptibility at the pas1 locus in kras2la2 mice. Nat. Genet. 2006, 38, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, Y.; Vikis, H.G.; Johnson, L.; Liu, G.; Li, J.; Anderson, M.W.; Sills, R.C.; Hong, H.L.; Devereux, T.R.; et al. Wildtype kras2 can inhibit lung carcinogenesis in mice. Nat. Genet. 2001, 29, 25–33. [Google Scholar] [CrossRef]
- Ambrogio, C.; Kohler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. Kras dimerization impacts mek inhibitor sensitivity and oncogenic activity of mutant kras. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef]
- Bentley, C.; Jurinka, S.S.; Kljavin, N.M.; Vartanian, S.; Ramani, S.R.; Gonzalez, L.C.; Yu, K.; Modrusan, Z.; Du, P.; Bourgon, R.; et al. A requirement for wild-type ras isoforms in mutant kras-driven signalling and transformation. Biochem. J. 2013, 452, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Mao, J.H.; Balmain, A. Allele-specific hras mutations and genetic alterations at tumor susceptibility loci in skin carcinomas from interspecific hybrid mice. Cancer Res. 2003, 63, 4849–4853. [Google Scholar] [PubMed]
- Estep, A.L.; Tidyman, W.E.; Teitell, M.A.; Cotter, P.D.; Rauen, K.A. Hras mutations in costello syndrome: Detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am. J. Med. Genet. A 2006, 140, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Bremner, R.; Balmain, A. Genetic changes in skin tumor progression: Correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell 1990, 61, 407–417. [Google Scholar] [CrossRef]
- Buchmann, A.; Bauer-Hofmann, R.; Mahr, J.; Drinkwater, N.R.; Luz, A.; Schwarz, M. Mutational activation of the c-ha-ras gene in liver tumors of different rodent strains: Correlation with susceptibility to hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 911–915. [Google Scholar] [CrossRef] [Green Version]
- Helias-Rodzewicz, Z.; Funck-Brentano, E.; Terrones, N.; Beauchet, A.; Zimmermann, U.; Marin, C.; Saiag, P.; Emile, J.F. Variation of mutant allele frequency in nras q61 mutated melanomas. BMC Dermatol. 2017, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Sahin, F.; Iacobuzio-Donahue, C.A.; Garcia-Carracedo, D.; Wang, W.M.; Kuo, C.Y.; Chen, D.; Arking, D.E.; Lowy, A.M.; Hruban, R.H.; et al. Disruption of p16 and activation of kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2011, 2, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.C.; Qiu, W.; Juang, C.S.; Mansukhani, M.M.; Halmos, B.; Su, G.H. Mutant allele specific imbalance in oncogenes with copy number alterations: Occurrence, mechanisms, and potential clinical implications. Cancer Lett. 2017, 384, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, Z.; Dai, Z.; Plass, C.; Morrison, C.; Wang, Y.; Wiest, J.S.; Anderson, M.W.; You, M. Loh of chromosome 12p correlates with kras2 mutation in non-small cell lung cancer. Oncogene 2003, 22, 1243–1246. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, I.; Villasante, A.; Corces, V.; Pellicer, A. Loss of the normal n-ras allele in a mouse thymic lymphoma induced by a chemical carcinogen. Proc. Natl. Acad. Sci. USA 1985, 82, 7810–7814. [Google Scholar] [CrossRef] [Green Version]
- Soh, J.; Okumura, N.; Lockwood, W.W.; Yamamoto, H.; Shigematsu, H.; Zhang, W.; Chari, R.; Shames, D.S.; Tang, X.; MacAulay, C.; et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (masi) frequently occur together in tumor cells. PLoS ONE 2009, 4, e7464. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. Kras allelic imbalance enhances fitness and modulates map kinase dependence in cancer. Cell 2017, 168, 817–829.e15. [Google Scholar] [CrossRef] [Green Version]
- Chiosea, S.I.; Sherer, C.K.; Jelic, T.; Dacic, S. Kras mutant allele-specific imbalance in lung adenocarcinoma. Mod. Pathol. 2011, 24, 1571–1577. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Makarewicz, J.M.; Knauf, J.A.; Johnson, L.K.; Fagin, J.A. Transformation by hras(g12v) is consistently associated with mutant allele copy gains and is reversed by farnesyl transferase inhibition. Oncogene 2014, 33, 5442–5449. [Google Scholar] [CrossRef] [Green Version]
- Bielski, C.M.; Donoghue, M.T.A.; Gadiya, M.; Hanrahan, A.J.; Won, H.H.; Chang, M.T.; Jonsson, P.; Penson, A.V.; Gorelick, A.; Harris, C.; et al. Widespread selection for oncogenic mutant allele imbalance in cancer. Cancer Cell 2018, 34, 852–862.e4. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Ollinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and kras dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Jang, H.; Gursoy, A.; Keskin, O.; Nussinov, R. Pdedelta binding to ras isoforms provides a route to proper membrane localization. J. Phys. Chem. B 2017, 121, 5917–5927. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; Jacobs, M.; Herrero-Garcia, E.; et al. Inhibition of ras function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef]
- Lin, W.C.; Iversen, L.; Tu, H.L.; Rhodes, C.; Christensen, S.M.; Iwig, J.S.; Hansen, S.D.; Huang, W.Y.; Groves, J.T. H-ras forms dimers on membrane surfaces via a protein-protein interface. Proc. Natl. Acad. Sci. USA 2014, 111, 2996–3001. [Google Scholar] [CrossRef] [Green Version]
- Guldenhaupt, J.; Rudack, T.; Bachler, P.; Mann, D.; Triola, G.; Waldmann, H.; Kotting, C.; Gerwert, K. N-ras forms dimers at popc membranes. Biophys. J. 2012, 103, 1585–1593. [Google Scholar] [CrossRef]
- Young, A.; Lou, D.; McCormick, F. Oncogenic and wild-type ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013, 3, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting biological insights from the project achilles genome-scale crispr screens in cancer cell lines. bioRxiv 2019, 720243. [Google Scholar]
- Yuan, T.L.; Amzallag, A.; Bagni, R.; Yi, M.; Afghani, S.; Burgan, W.; Fer, N.; Strathern, L.A.; Powell, K.; Smith, B.; et al. Differential Effector Engagement by Oncogenic KRAS. Cell Rep. 2018, 22, 1889–1902. [Google Scholar] [CrossRef] [Green Version]
- Balbin, O.A.; Prensner, J.R.; Sahu, A.; Yocum, A.K.; Shankar, S.; Malik, R.; Fermin, D.; Dhanasekaran, S.M.; Chandler, B.; Thomas, D.G.; et al. Reconstructing targetable pathways in lung cancer by integrating diverse omics data. Nat. Commun. 2013, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Sweeney, M.F.; Yu, M.; Burger, A.; Greninger, P.; Benes, C.; Haber, D.A.; Settleman, J. TAK1 Inhibition Promotes Apoptosis in KRAS-Dependent Colon Cancers. Cell 2012, 148, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Scholl, C.; Fröhling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic Lethal Interaction between Oncogenic KRAS Dependency and STK33 Suppression in Human Cancer Cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef] [Green Version]
- Lamba, S.; Russo, M.; Sun, C.; Lazzari, L.; Cancelliere, C.; Grernrum, W.; Lieftink, C.; Bernards, R.; Di Nicolantonio, F.; Bardelli, A. RAF Suppression Synergizes with MEK Inhibition in KRAS Mutant Cancer Cells. Cell Rep. 2014, 8, 1475–1483. [Google Scholar] [CrossRef] [Green Version]
- Fujita-Sato, S.; Galeas, J.; Truitt, M.; Pitt, C.; Urisman, A.; Bandyopadhyay, S.; Ruggero, D.; McCormick, F. Enhanced MET Translation and Signaling Sustains K-Ras–Driven Proliferation under Anchorage-Independent Growth Conditions. Cancer Res. 2015, 75, 2851–2862. [Google Scholar] [CrossRef] [Green Version]
- Rotem, A.; Janzer, A.; Izar, B.; Ji, Z.; Doench, J.G.; Garraway, L.A.; Struhl, K. Alternative to the soft-agar assay that permits high-throughput drug and genetic screens for cellular transformation. Proc. Natl. Acad. Sci. USA 2015, 112, 5708–5713. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Jiang, G.; Yang, F.; Wang, J. Knockdown of mutant K-ras expression by adenovirus-mediated siRNA inhibits the in vitro and in vivo growth of lung cancer cells. Cancer Biol. Ther. 2006, 5, 1481–1486. [Google Scholar] [CrossRef] [Green Version]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [Green Version]
- Margarit, S.; Sondermann, H.; Hall, B.E.; Nagar, B.; Hoelz, A.; Pirruccello, M.; Bar-Sagi, D.; Kuriyan, J. Structural Evidence for Feedback Activation by Ras·GTP of the Ras-Specific Nucleotide Exchange Factor SOS. Cell 2003, 112, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, H.; Soisson, S.M.; Boykevisch, S.; Yang, S.-S.; Bar-Sagi, D.; Kuriyan, J. Structural Analysis of Autoinhibition in the Ras Activator Son of Sevenless. Cell 2004, 119, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Boykevisch, S.; Zhao, C.; Sondermann, H.; Philippidou, P.; Halegoua, S.; Kuriyan, J.; Bar-Sagi, D. Regulation of Ras Signaling Dynamics by Sos-Mediated Positive Feedback. Curr. Biol. 2006, 16, 2173–2179. [Google Scholar] [CrossRef] [Green Version]
- Iversen, L.F.; Tu, H.-L.; Lin, W.-C.; Christensen, S.M.; Abel, S.M.; Iwig, J.S.; Wu, H.-J.; Gureasko, J.M.; Rhodes, C.P.; Petit, R.S.; et al. Ras activation by SOS: Allosteric regulation by altered fluctuation dynamics. Science 2014, 345, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, S.M.; Tu, H.-L.; Jun, J.E.; Alvarez, S.; Triplet, M.G.; Iwig, J.S.; Yadav, K.K.; Bar-Sagi, D.; Roose, J.P.; Groves, J.T. One-way membrane trafficking of SOS in receptor-triggered Ras activation. Nat. Struct. Mol. Biol. 2016, 23, 838–846. [Google Scholar] [CrossRef]
- Huang, W.Y.C.; Alvarez, S.; Kondo, Y.; Lee, Y.K.; Chung, J.K.; Lam, H.Y.M.; Biswas, K.H.; Kuriyan, J.; Groves, J.T. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 2019, 363, 1098–1103. [Google Scholar] [CrossRef]
- Tartaglia, M.; Pennacchio, L.A.; Zhao, C.; Yadav, K.K.; Fodale, V.; Sarkozy, A.; Pandit, B.; Oishi, K.; Martinelli, S.; Schackwitz, W.; et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2006, 39, 75–79. [Google Scholar] [CrossRef]
- Umutesi, H.G.; Hoang, H.M.; Johnson, H.E.; Nam, K.; Heo, J. Development of Noonan syndrome by deregulation of allosteric SOS autoactivation. J. Biol. Chem. 2020, 295, 13651–13663. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Zikherman, J.; Das, J.; Roose, J.P.; Weiss, A.; Chakraborty, A.K. Origin of the sharp boundary that discriminates positive and negative selection of thymocytes. Proc. Natl. Acad. Sci. USA 2008, 106, 528–533. [Google Scholar] [CrossRef] [Green Version]
- Daniels, M.A.; Teixeiro, E.; Gill, J.; Hausmann, B.; Roubaty, D.; Holmberg, K.; Werlen, G.; Holländer, G.A.; Gascoigne, N.R.J.; Palmer, E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nat. Cell Biol. 2006, 444, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Kortum, R.L.; Sommers, C.L.; Pinski, J.M.; Alexander, C.P.; Merrill, R.K.; Li, W.; Love, P.E.; Samelson, L.E. Deconstructing Ras Signaling in the Thymus. Mol. Cell. Biol. 2012, 32, 2748–2759. [Google Scholar] [CrossRef] [Green Version]
- Roose, J.P.; Mollenauer, M.; Ho, M.; Kurosaki, T.; Weiss, A. Unusual Interplay of Two Types of Ras Activators, RasGRP and SOS, Establishes Sensitive and Robust Ras Activation in Lymphocytes. Mol. Cell. Biol. 2007, 27, 2732–2745. [Google Scholar] [CrossRef] [Green Version]
- Das, J.; Ho, M.; Zikherman, J.; Govern, C.; Yang, M.; Weiss, A.; Chakraborty, A.K.; Roose, J.P. Digital Signaling and Hysteresis Characterize Ras Activation in Lymphoid Cells. Cell 2009, 136, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Jeng, H.-H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, M.; Wolfman, A. Oncogenic Ha-Ras-dependent Mitogen-activated Protein Kinase Activity Requires Signaling through the Epidermal Growth Factor Receptor. J. Biol. Chem. 1998, 273, 28155–28162. [Google Scholar] [CrossRef] [Green Version]
- Grabocka, E.; Pylayeva-Gupta, Y.; Jones, M.J.; Lubkov, V.; Yemanaberhan, E.; Taylor, L.; Jeng, H.H.; Bar-Sagi, D. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell 2014, 25, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.-H.; Ancrile, B.B.; Kashatus, D.F.; Counter, C.M. Tumour maintenance is mediated by eNOS. Nat. Cell Biol. 2008, 452, 646–649. [Google Scholar] [CrossRef] [Green Version]
- Sheffels, E.; Sealover, N.E.; Wang, C.; Kim, D.H.; Vazirani, I.A.; Lee, E.; Terrell, E.M.; Morrison, D.K.; Luo, J.; Kortum, R.L. Oncogenic RAS isoforms show a hierarchical requirement for the guanine nucleotide exchange factor SOS2 to mediate cell transformation. Sci. Signal. 2018, 11, eaar8371. [Google Scholar] [CrossRef] [Green Version]
- Sheffels, E.; Sealover, N.E.; Theard, P.L.; Kortum, R.L. Anchorage-independent growth conditions reveal a differential SOS2 dependence for transformation and survival in RAS-mutant cancer cells. Small GTPases 2021, 12, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Roy, S.; Apolloni, A.; Lane, A.; Hancock, J.F. Ras Isoforms Vary in Their Ability to Activate Raf-1 and Phosphoinositide 3-Kinase. J. Biol. Chem. 1998, 273, 24052–24056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voice, J.K.; Klemke, R.L.; Le, A.; Jackson, J.H. Four Human Ras Homologs Differ in Their Abilities to Activate Raf-1, Induce Transformation, and Stimulate Cell Motility. J. Biol. Chem. 1999, 274, 17164–17170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, C.; Martin, G.A.; Wittinghofer, A. Quantitative Analysis of the Complex between p21 and the Ras-binding Domain of the Human Raf-1 Protein Kinase. J. Biol. Chem. 1995, 270, 2901–2905. [Google Scholar] [CrossRef] [Green Version]
- Maher, J.; Baker, D.A.; Manning, M.; Dibb, N.J.; Roberts, I.A. Evidence for cell-specific differences in transformation by N-, H- and K-ras. Oncogene 1995, 11, 1639–1647. [Google Scholar] [PubMed]
- Terrell, E.M.; Durrant, D.E.; Ritt, D.A.; Sealover, N.E.; Sheffels, E.; Spencer-Smith, R.; Esposito, D.; Zhou, Y.; Hancock, J.F.; Kortum, R.L.; et al. Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol. Cell 2019, 76, 872–884.e5. [Google Scholar] [CrossRef]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. Effects of raf dimerization and its inhibition on normal and disease-associated raf signaling. Mol. Cell 2013, 49, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Prakash, P.; Liang, H.; Cho, K.-J.; Gorfe, A.A.; Hancock, J.F. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell 2017, 168, 239–251.e16. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Santos, E. Functional Specificity of Ras Isoforms: So Similar but So Different. Genes Cancer 2011, 2, 216–231. [Google Scholar] [CrossRef] [Green Version]
- Untch, B.R.; Dos Anjos, V.; Garcia-Rendueles, M.E.R.; Knauf, J.A.; Krishnamoorthy, G.P.; Saqcena, M.; Bhanot, U.K.; Socci, N.D.; Ho, A.L.; Ghossein, R.; et al. Tipifarnib Inhibits HRAS-Driven Dedifferentiated Thyroid Cancers. Cancer Res. 2018, 78, 4642–4657. [Google Scholar] [CrossRef] [Green Version]
- Ebi, H.; Corcoran, R.B.; Singh, A.; Chen, Z.; Song, Y.; Lifshits, E.; Ryan, D.P.; Meyerhardt, J.A.; Benes, C.; Settleman, J.; et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J. Clin. Investig. 2011, 121, 4311–4321. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Hancock, D.C.; Sheridan, C.; Kumar, M.S.; Downward, J. Coordinate Direct Input of Both KRAS and IGF1 Receptor to Activation of PI3 kinase in KRAS-Mutant Lung Cancer. Cancer Discov. 2013, 3, 548–563. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK Inhibition Leads to PI3K/AKT Activation by Relieving a Negative Feedback on ERBB Receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [Green Version]
- Pettazzoni, P.; Viale, A.; Shah, P.; Carugo, A.; Ying, H.; Wang, H.; Genovese, G.; Seth, S.; Minelli, R.; Green, T.; et al. Genetic Events That Limit the Efficacy of MEK and RTK Inhibitor Therapies in a Mouse Model of KRAS-Driven Pancreatic Cancer. Cancer Res. 2015, 75, 1091–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sos, M.L.; Fischer, S.; Ullrich, R.; Peifer, M.; Heuckmann, J.M.; Koker, M.; Heynck, S.; Stückrath, I.; Weiss, J.; Fischer, F.; et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 18351–18356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yohe, M.E.; Gryder, B.E.; Shern, J.F.; Song, Y.K.; Chou, H.-C.; Sindiri, S.; Mendoza, A.; Patidar, R.; Zhang, X.; Guha, R.; et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018, 10, eaan4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.A.; Liu, Y.; Zage, P.E. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef] [Green Version]
- Jessen, W.J.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.O.; Dombi, E.; et al. Mek inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Investig. 2013, 123, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Malek, S. Targeting the mapk pathway in ras mutant cancers. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target ras-mutant cancers. Nat. Rev. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic resistance to mek inhibition in kras mutant lung and colon cancer through transcriptional induction of erbb3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchado, E.; Weissmueller, S.; Morris, J.P.t.; Chen, C.C.; Wullenkord, R.; Lujambio, A.; de Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating kras-mutant lung cancer. Nature 2016, 534, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Chandarlapaty, S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov. 2012, 2, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, A.; Sanchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of her3 (erbb3) expression and activity attenuates antitumor effect of pi3k inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 2718–2723. [Google Scholar] [CrossRef] [Green Version]
- Renshaw, J.; Taylor, K.R.; Bishop, R.; Valenti, M.; Brandon, A.d.; Gowan, S.; Eccles, S.A.; Ruddle, R.R.; Johnson, L.D.; Raynaud, F.I.; et al. Dual blockade of the pi3k/akt/mtor (azd8055) and ras/mek/erk (azd6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clin. Cancer Res. 2013, 19, 5940–5951. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of pi3k and mek inhibitors to treat mutant k-ras g12d and pik3ca h1047r murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving pi3k/akt/mtor and ras/mek/erk pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [Green Version]
- Jokinen, E.; Koivunen, J.P. Mek and pi3k inhibition in solid tumors: Rationale and evidence to date. Ther. Adv. Med. Oncol. 2015, 7, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.R.; Winter, P.S.; Lin, K.H.; Nussbaum, D.P.; Cakir, M.; Stein, E.M.; Soderquist, R.S.; Crawford, L.; Leeds, J.C.; Newcomb, R.; et al. A landscape of therapeutic cooperativity in kras mutant cancers reveals principles for controlling tumor evolution. Cell Rep. 2017, 20, 999–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. Shp2 inhibition prevents adaptive resistance to mek inhibitors in multiple cancer models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. Shp2 inhibition diminishes krasg12c cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Vitiello, M.; Palma, G.; Monaco, M.; Bello, A.M.; Camorani, S.; Francesca, P.; Rea, D.; Barbieri, A.; Chiappetta, G.; Vita, G.; et al. Dual oncogenic/anti-oncogenic role of patz1 in frtl5 rat thyroid cells transformed by the ha-ras(v12) oncogene. Genes 2019, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Rowell, C.A.; Kowalczyk, J.J.; Lewis, M.D.; Garcia, A.M. Direct demonstration of geranylgeranylation and farnesylation of ki-ras in vivo. J. Biol. Chem. 1997, 272, 14093–14097. [Google Scholar] [CrossRef] [Green Version]
- James, G.L.; Goldstein, J.L.; Brown, M.S. Polylysine and cvim sequences of k-rasb dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J. Biol. Chem. 1995, 270, 6221–6226. [Google Scholar] [CrossRef] [Green Version]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and n-ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Cunningham, D.; de Gramont, A.; Scheithauer, W.; Smakal, M.; Humblet, Y.; Kourteva, G.; Iveson, T.; Andre, T.; Dostalova, J.; et al. Phase iii double-blind placebo-controlled study of farnesyl transferase inhibitor r115777 in patients with refractory advanced colorectal cancer. J. Clin. Oncol. 2004, 22, 3950–3957. [Google Scholar] [CrossRef]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase iii trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting ras membrane association: Back to the future for anti-ras drug discovery? Clin. Cancer Res 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, A.; Chau, N.; Garcia, I.B.; Ferte, C.; Even, C.; Burrows, F.; Kessler, L.; Mishra, V.; Magnuson, K.; Scholz, C.; et al. Preliminary results from a phase 2 trial of tipifarnib in hras-mutant head and neck squamous cell carcinomas. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1367. [Google Scholar] [CrossRef]
- Lee, H.W.; Sa, J.K.; Gualberto, A.; Scholz, C.; Sung, H.H.; Jeong, B.C.; Choi, H.Y.; Kwon, G.Y.; Park, S.H. A phase ii trial of tipifarnib for patients with previously treated, metastatic urothelial carcinoma harboring hras mutations. Clin. Cancer Res. 2020, 26, 5113–5119. [Google Scholar] [CrossRef]
- Hanna, G.J.; Guenette, J.P.; Chau, N.G.; Sayehli, C.M.; Wilhelm, C.; Metcalf, R.; Wong, D.J.; Brose, M.; Razaq, M.; Perez-Ruiz, E.; et al. Tipifarnib in recurrent, metastatic hras-mutant salivary gland cancer. Cancer 2020, 126, 3972–3981. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; de la Cruz, F.F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to kras(g12c) inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific kras(g12c) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. Kras(g12c) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal 2019, 12. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. Kras g12c nsclc models are sensitive to direct targeting of kras in combination with pi3k inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of kras-g12c inhibitors in lung cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Puyol, M.; Martin, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaria, D.; Barbacid, M. A synthetic lethal interaction between k-ras oncogenes and cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of shp2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Garcia Fortanet, J.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric inhibition of shp2: Identification of a potent, selective, and orally efficacious phosphatase inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. Ras nucleotide cycling underlies the shp2 phosphatase dependence of mutant braf-, nf1- and ras-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent sos1 inhibitors that block ras activation via disruption of the ras-sos1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. Bi-3406, a potent and selective sos1::Kras interaction inhibitor, is effective in kras-driven cancers through combined mek inhibition. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Ramharter, J.; Kessler, D.; Ettmayer, P.; Hofmann, M.H.; Gerstberger, T.; Gmachl, M.; Wunberg, T.; Kofink, C.; Sanderson, M.; Arnhof, H.; et al. One atom makes all the difference: Getting a foot in the door between sos1 and kras. J. Med. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Gorgulu, K.; Dantes, Z.; Wormann, S.M.; Diakopoulos, K.N.; et al. Mutant kras-driven cancers depend on ptpn11/shp2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sama, I.; Ladumor, Y.; Kee, L.; Adderley, T.; Christopher, G.; Robinson, C.M.; Kano, Y.; Ohh, M.; Irwin, M.S. Nras status determines sensitivity to shp2 inhibitor combination therapies targeting the ras-mapk pathway in neuroblastoma. Cancer Res. 2020, 80, 3413–3423. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Loupakis, F.; Ruzzo, A.; Cremolini, C.; Vincenzi, B.; Salvatore, L.; Santini, D.; Masi, G.; Stasi, I.; Canestrari, E.; Rulli, E.; et al. Kras codon 61, 146 and braf mutations predict resistance to cetuximab plus irinotecan in kras codon 12 and 13 wild-type metastatic colorectal cancer. Br. J. Cancer 2009, 101, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-folfox4 treatment and ras mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Pinto, C.; Normanno, N.; Orlandi, A.; Fenizia, F.; Damato, A.; Maiello, E.; Tamburini, E.; di Costanzo, F.; Tonini, G.; Bilancia, D.; et al. Phase iii study with folfiri + cetuximab versus folfiri + cetuximab followed by cetuximab alone in ras and braf wt mcrc. Future Oncol. 2018, 14, 1339–1346. [Google Scholar] [CrossRef]
- Peeters, M.; Douillard, J.Y.; van Cutsem, E.; Siena, S.; Zhang, K.; Williams, R.; Wiezorek, J. Mutant kras codon 12 and 13 alleles in patients with metastatic colorectal cancer: Assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol. 2013, 31, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costas-Chavarri, A.; Nandakumar, G.; Temin, S.; Lopes, G.; Cervantes, A.; Correa, M.C.; Engineer, R.; Hamashima, C.; Ho, G.F.; Huitzil, F.D.; et al. Treatment of patients with early-stage colorectal cancer: Asco resource-stratified guideline. J. Glob. Oncol. 2019, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Jonker, D.J.; di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of kras p.G13d mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segelov, E.; Thavaneswaran, S.; Waring, P.M.; Desai, J.; Robledo, K.P.; Gebski, V.J.; Elez, E.; Nott, L.M.; Karapetis, C.S.; Lunke, S.; et al. Response to cetuximab with or without irinotecan in patients with refractory metastatic colorectal cancer harboring the kras g13d mutation: Australasian gastro-intestinal trials group icecream study. J. Clin. Oncol. 2016, 34, 2258–2264. [Google Scholar] [CrossRef]
- Segelov, E.; Waring, P.; Desai, J.; Wilson, K.; Gebski, V.; Thavaneswaran, S.; Elez, E.; Underhill, C.; Pavlakis, N.; Chantrill, L.; et al. Icecream: Randomised phase ii study of cetuximab alone or in combination with irinotecan in patients with metastatic colorectal cancer with either kras, nras, braf and pi3kca wild type, or g13d mutated tumours. BMC Cancer 2016, 16, 339. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Aoyama, T.; Ishibashi, K.; Tsuji, A.; Takinishi, Y.; Shindo, Y.; Sakamoto, J.; Oba, K.; Mishima, H. Randomized phase ii study of cetuximab versus irinotecan and cetuximab in patients with chemo-refractory kras codon g13d metastatic colorectal cancer (g13d-study). Cancer Chemother. Pharmacol. 2017, 79, 29–36. [Google Scholar] [CrossRef] [Green Version]
- McFall, T.; Diedrich, J.K.; Mengistu, M.; Littlechild, S.L.; Paskvan, K.V.; Sisk-Hackworth, L.; Moresco, J.J.; Shaw, A.S.; Stites, E.C. A systems mechanism for kras mutant allele-specific responses to targeted therapy. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, H.T.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical kras(g12r) mutant is impaired in pi3k signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nat. Cell Biol. 2013, 497, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Heiden, M.G.V.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Zafra, M.P.; Parsons, M.J.; Kim, J.; Alonso-Curbelo, D.; Goswami, S.; Schatoff, E.M.; Han, T.; Katti, A.; Fernandez, M.T.C.; Wilkinson, J.E.; et al. An In Vivo Kras Allelic Series Reveals Distinct Phenotypes of Common Oncogenic Variants. Cancer Discov. 2020, 10, 1654–1671. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheffels, E.; Kortum, R.L. The Role of Wild-Type RAS in Oncogenic RAS Transformation. Genes 2021, 12, 662. https://doi.org/10.3390/genes12050662
Sheffels E, Kortum RL. The Role of Wild-Type RAS in Oncogenic RAS Transformation. Genes. 2021; 12(5):662. https://doi.org/10.3390/genes12050662
Chicago/Turabian StyleSheffels, Erin, and Robert L. Kortum. 2021. "The Role of Wild-Type RAS in Oncogenic RAS Transformation" Genes 12, no. 5: 662. https://doi.org/10.3390/genes12050662
APA StyleSheffels, E., & Kortum, R. L. (2021). The Role of Wild-Type RAS in Oncogenic RAS Transformation. Genes, 12(5), 662. https://doi.org/10.3390/genes12050662