
The Molecular Basis of Calcium and Phosphorus Inherited Metabolic Disorders

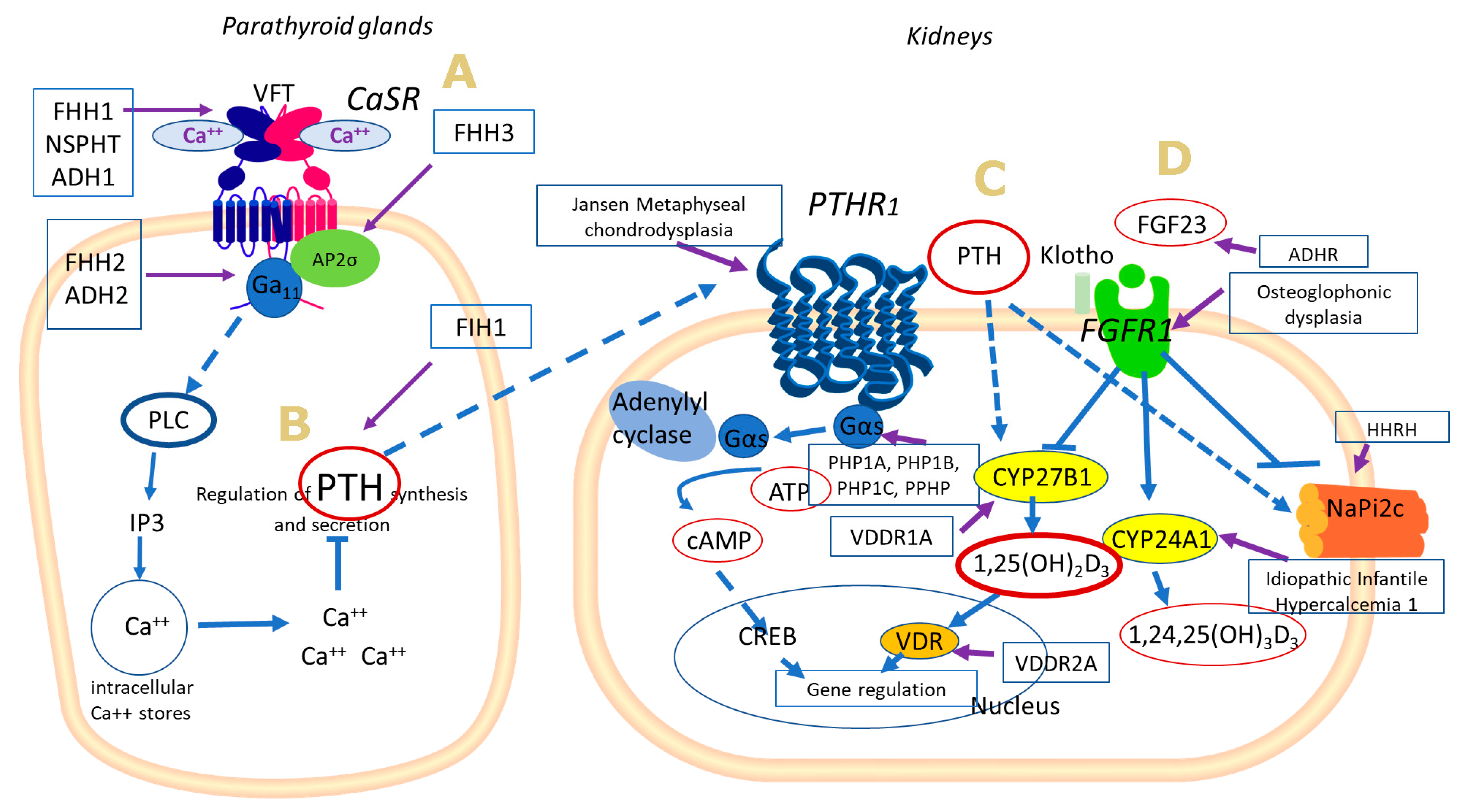
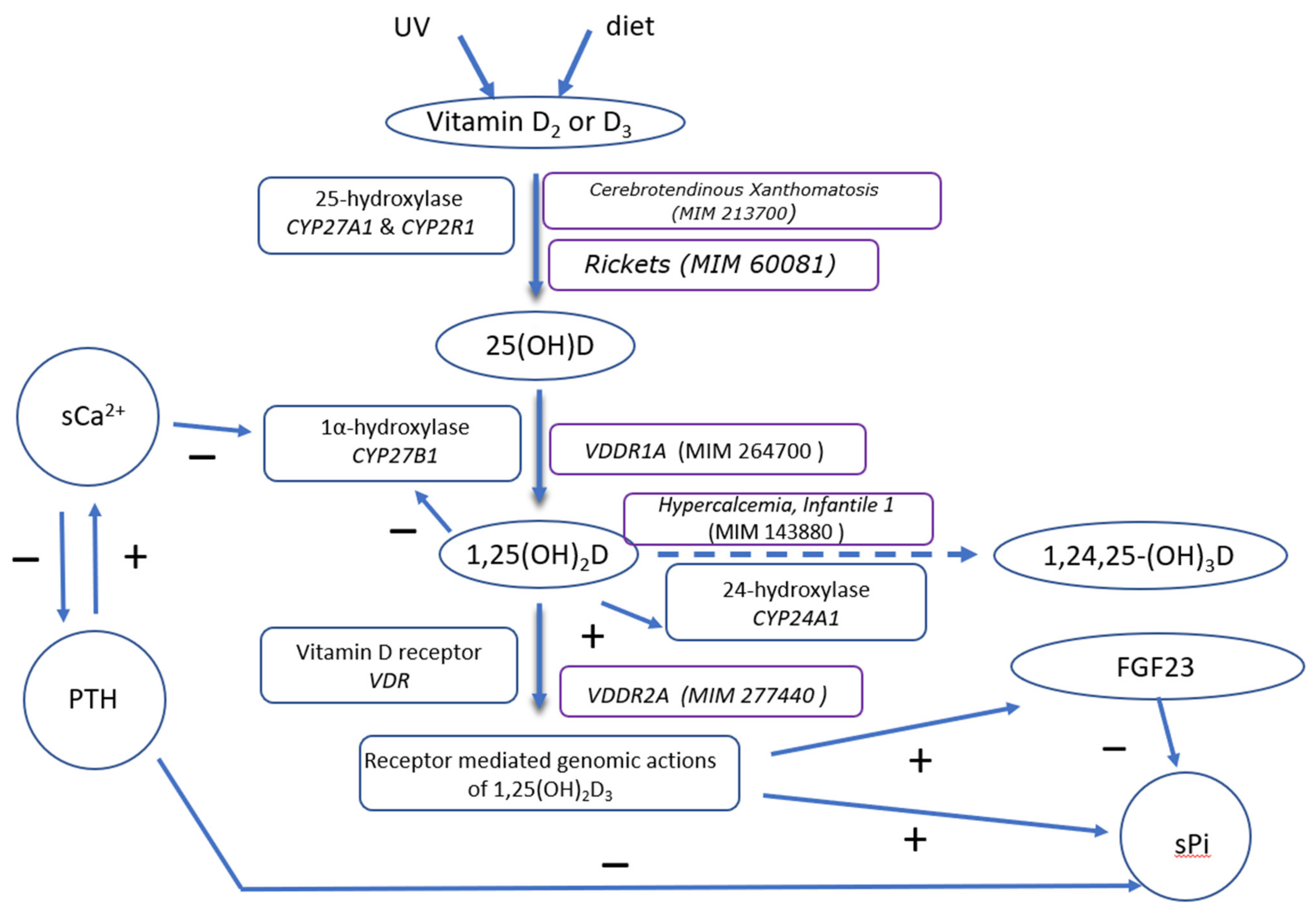
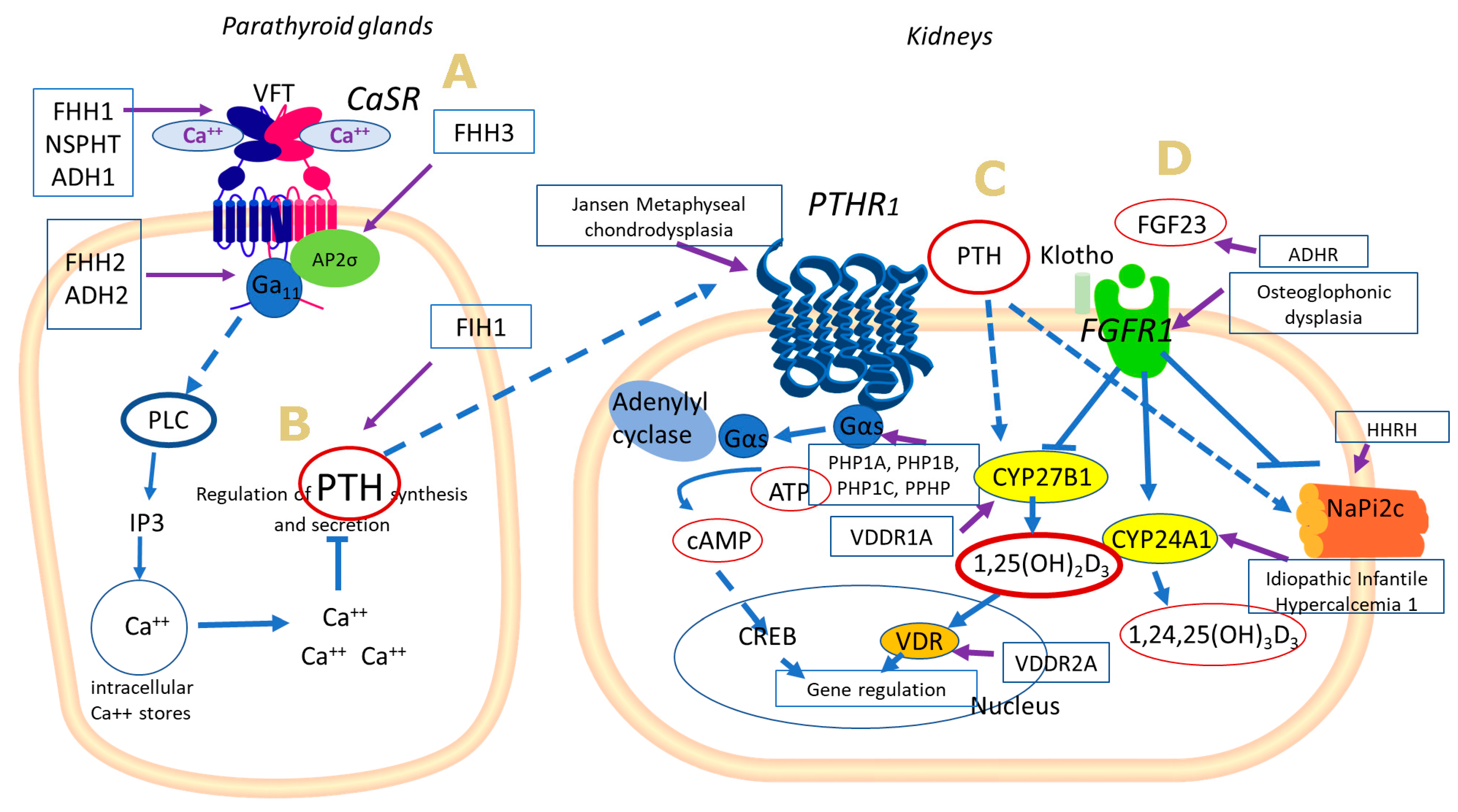{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Calcium and Its Partners
2.1. Calcium Sensing Receptor
2.2. Parathyroid Hormone
2.3. Vitamin D
2.4. Inherited Diseases Associated with Defective Extracellular Ca Sensing Mechanism
2.4.1. Familial Hypocalciuric Hypercalcaemia
2.4.2. Autosomal Dominant Hypocalcaemia
2.5. Inherited Diseases Associated with Defective PTH Synthesis and Action
2.5.1. Familial Isolated Hypoparathyroidism, Type 1
2.5.2. Jansen Metaphyseal Chondrodysplasia
2.5.3. Pseudohypoparathyroidism
2.6. Inherited Disorders Associated with Defective Vitamin D Metabolism
2.6.1. Vitamin D Hydroxylation Deficient Rickets, Type 1A, or Pseudovitamin D Deficiency Rickets, or 1α-Hydroxylase Deficiency
2.6.2. Vitamin D-Dependent Rickets Type 2A
3. Phosphorus and Its Partners
3.1. Hypophosphatemic Rickets with Normal or Decreased FGF23 Levels
3.2. Hypophosphatemic Rickets with Elevated FGF23 Levels
3.2.1. X-Linked Hypophosphatemic Rickets (XLH)
3.2.2. Autosomal Dominant Hypophosphatemic Rickets
3.2.3. Diseases Associated with FGFR1 Gene Mutations
3.2.4. Autosomal Recessive Hypophosphatemic Rickets
3.2.5. Diseases Associated with Klotho Gene Mutations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Peacock, M. Calcium metabolism in health and disease. Clin. J. Am. Soc. Nephrol. 2010, 5 (Suppl. 1), S23–S30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, S.M. Calcium Homeostasis in Health and in Kidney Disease. Compr. Physiol. 2016, 6, 1781–1800. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.W. Ionized calcium in normal serum, ultrafiltrates, and whole blood determined by ion-exchange electrodes. J. Clin. Investig. 1970, 49, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Gattineni, J. Inherited disorders of calcium and phosphate metabolism. Curr. Opin. Pediatr. 2014, 26, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Goltzman, D.; Mannstadt, M.; Marcocci, C. Physiology of the Calcium-Parathyroid Hormone-Vitamin D Axis. Front. Horm. Res. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Friedman, P.A.; Gesek, F.A. Cellular calcium transport in renal epithelia: Measurement, mechanisms, and regulation. Physiol. Rev. 1995, 75, 429–471. [Google Scholar] [CrossRef]
- Hannan, F.M.; Kallay, E.; Chang, W.; Brandi, M.L.; Thakker, R.V. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 2018, 15, 33–51. [Google Scholar] [CrossRef]
- Geng, Y.; Mosyak, L.; Kurinov, I.; Zuo, H.; Sturchler, E.; Cheng, T.C.; Subramanyam, P.; Brown, A.P.; Brennan, S.C.; Mun, H.C.; et al. Structural mechanism of ligand activation in human calcium-sensing receptor. eLife 2016, 5, e13662. [Google Scholar] [CrossRef]
- Hannan, F.M.; Babinsky, V.N.; Thakker, R.V. Disorders of the calcium-sensing receptor and partner proteins: Insights into the molecular basis of calcium homeostasis. J. Mol. Endocrinol. 2016, 57, R127–R142. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Zhou, Y.; Yang, W.; Butters, R.; Lee, H.-W.; Li, S.; Castiblanco, A.; Brown, E.M.; Yang, J.J. Identification and Dissection of Ca2+-binding Sites in the Extracellular Domain of Ca2+-sensing Receptor. J. Biol. Chem. 2007, 282, 19000–19010. [Google Scholar] [CrossRef] [Green Version]
- Conigrave, A.D.; Ward, D.T. Calcium-sensing receptor (CaSR): Pharmacological properties and signaling pathways. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 315–331. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, T.; Zou, J.; Miller, C.L.; Gorkhali, R.; Yang, J.Y.; Schilmiller, A.; Wang, S.; Huang, K.; Brown, E.M.; et al. Structural basis for regulation of human calcium-sensing receptor by magnesium ions and an unexpected tryptophan derivative co-agonist. Sci. Adv. 2016, 2, e1600241. [Google Scholar] [CrossRef] [Green Version]
- Conigrave, A.D. The Calcium-Sensing Receptor and the Parathyroid: Past, Present, Future. Front. Physiol. 2016, 7, 563. [Google Scholar] [CrossRef]
- Hjalm, G.; MacLeod, R.J.; Kifor, O.; Chattopadhyay, N.; Brown, E.M. Filamin-A binds to the carboxyl-terminal tail of the calcium-sensing receptor, an interaction that participates in CaR-mediated activation of mitogen-activated protein kinase. J. Biol. Chem. 2001, 276, 34880–34887. [Google Scholar] [CrossRef] [Green Version]
- Kifor, O.; Kifor, I.; Moore, F.D.; Butters, R.R.; Cantor, T.; Gao, P.; Brown, E.M. Decreased Expression of Caveolin-1 and Altered Regulation of Mitogen-Activated Protein Kinase in Cultured Bovine Parathyroid Cells and Human Parathyroid Adenomas. J. Clin. Endocrinol. Metab. 2003, 88, 4455–4464. [Google Scholar] [CrossRef] [Green Version]
- Goodman, W.G.; Veldhuis, J.D.; Belin, T.R.; van Herle, A.J.; Juppner, H.; Salusky, I.B. Calcium-Sensing by Parathyroid Glands in Secondary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 1998, 83, 2765–2772. [Google Scholar] [CrossRef]
- Gordon, R.J.; Levine, M.A. Genetic Disorders of Parathyroid Development and Function. Endocrinol. Metab. Clin. N. Am. 2018, 47, 809–823. [Google Scholar] [CrossRef]
- Brown, E.M. Physiology of calcium homeostasis. In The Parathyroids; Bilezikian, J.P., Marcus, R., Levine, A., Eds.; Academic Press: Cambridge, MA, USA, 2001; pp. 167–182. [Google Scholar]
- Shaker, J.L.; Deftos, L. Calcium and Phosphate Homeostasis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Cheloha, R.W.; Gellman, S.H.; Vilardaga, J.P.; Gardella, T.J. PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat. Rev. Endocrinol. 2015, 11, 712–724. [Google Scholar] [CrossRef] [Green Version]
- Goltzman, D. Studies on the mechanisms of the skeletal anabolic action of endogenous and exogenous parathyroid hormone. Arch. Biochem. Biophys. 2008, 473, 218–224. [Google Scholar] [CrossRef]
- Haddad, J.G. Vitamin D—Solar rays, the Milky Way, or both? N. Engl. J. Med. 1992, 326, 1213–1215. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D: A millenium perspective. J. Cell. Biochem. 2002, 88, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Hossein-Nezhad, A.; Holick, M.F. Vitamin D for Health: A Global Perspective. Mayo Clin. Proc. 2013, 88, 720–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allgrove, J. Is nutritional rickets returning? Arch. Dis. Child 2004, 89, 699–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaidou, P.; Hatzistamatiou, Z.; Papadopoulou, A.; Kaleyias, J.; Floropoulou, E.; Lagona, E.; Tsagris, V.; Costalos, C.; Antsaklis, A. Low Vitamin D Status in Mother-Newborn Pairs in Greece. Calcif. Tissue Int. 2006, 78, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Bountouvi, E.; Papaevaggelou, V.; Priftis, K.N. Maternal Vitamin D Status and Development of Asthma and Allergy in Early Childhood. Mini Rev. Med. Chem. 2015, 15, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.G.; Ochalek, J.T.; Kaufmann, M.; Jones, G.; DeLuca, H.F. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 15650–15655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, J.J.; Hsieh, C.L.; Francke, U.; Russell, D.W. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J. Biol. Chem. 1991, 266, 7779–7783. [Google Scholar] [CrossRef]
- Thacher, T.D.; Fischer, P.R.; Singh, R.J.; Roizen, J.; Levine, M.A. CYP2R1 Mutations Impair Generation of 25-hydroxyvitamin D and Cause an Atypical Form of Vitamin D Deficiency. J. Clin. Endocrinol. Metab. 2015, 100, E1005–E1013. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, Treatment, and Prevention of Vitamin D Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D. Extra Renal Synthesis of 1,25-dihydroxyvitamin D and its Health Implications. Clin. Rev. Bone Miner. Metab. 2009, 7, 114–125. [Google Scholar] [CrossRef]
- St-Arnaud, R.; Messerlian, S.; Moir, J.M.; Omdahl, J.L.; Glorieux, F.H. The 25-Hydroxyvitamin D 1-α-Hydroxylase Gene Maps to the Pseudovitamin D-Deficiency Rickets (PDDR) Disease Locus. J. Bone Miner. Res. 1997, 12, 1552–1559. [Google Scholar] [CrossRef]
- Bikle, D. Vitamin D: Production, Metabolism, and Mechanisms of Action. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Xie, Z.; Munson, S.J.; Huang, N.; Portale, A.A.; Miller, W.L.; Bikle, D.D. The Mechanism of 1,25-Dihydroxyvitamin D3Autoregulation in Keratinocytes. J. Biol. Chem. 2002, 277, 36987–36990. [Google Scholar] [CrossRef] [Green Version]
- Schlingmann, K.P.; Kaufmann, M.; Weber, S.; Irwin, A.; Goos, C.; John, U.; Misselwitz, J.; Klaus, G.; Kuwertz-Bröking, E.; Fehrenbach, H.; et al. Mutations in CYP24A1and Idiopathic Infantile Hypercalcemia. N. Engl. J. Med. 2011, 365, 410–421. [Google Scholar] [CrossRef]
- Malloy, P.J.; Pike, J.W.; Feldman, D. The Vitamin D Receptor and the Syndrome of Hereditary 1,25-Dihydroxyvitamin D-Resistant Rickets. Endocr. Rev. 1999, 20, 156–188. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Strugnell, S.A.; DeLuca, H.F. Current understanding of the molecular actions of vitamin D. Physiol. Rev. 1998, 78, 1193–1231. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, C.; Seuter, S.; Heikkinen, S. The first genome-wide view of vitamin D receptor locations and their mechanistic implications. Anticancer Res. 2012, 32, 271–282. [Google Scholar]
- Hossein-Nezhad, A.; Spira, A.; Holick, M.F. Influence of Vitamin D Status and Vitamin D3 Supplementation on Genome Wide Expression of White Blood Cells: A Randomized Double-Blind Clinical Trial. PLoS ONE 2013, 8, e58725. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, R.H.; Kallfelz, F.A.; Comar, C.L. Active transport of calcium by rat duodenum in vivo. Science 1961, 133, 883–884. [Google Scholar] [CrossRef]
- Kumar, R.; Schaefer, J.; Grande, J.P.; Roche, P.C. Immunolocalization of calcitriol receptor, 24-hydroxylase cytochrome P-450, and calbindin D28k in human kidney. Am. J. Physiol. 1994, 266, F477–F485. [Google Scholar] [CrossRef]
- Bikle, D.D.; Bouillon, R. Vitamin D and bone and beyond. Bone Rep. 2018, 9, 120–121. [Google Scholar] [CrossRef]
- Garrett, J.E.; Capuano, I.V.; Hammerland, L.G.; Hung, B.C.; Brown, E.M.; Hebert, S.C.; Nemeth, E.F.; Fuller, F. Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. J. Biol. Chem. 1995, 270, 12919–12925. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.E.; Nissen, P.H.; Vestergaard, P.; Heickendorff, L.; Rejnmark, L.; Brixen, K.; Mosekilde, L. Plasma 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and parathyroid hormone in familial hypocalciuric hypercalcemia and primary hyperparathyroidism. Eur. J. Endocrinol. 2008, 159, 719–727. [Google Scholar] [CrossRef]
- Gorvin, C.M.; Metpally, R.; Stokes, V.J.; Hannan, F.M.; Krishnamurthy, S.B.; Overton, J.D.; Reid, J.G.; Breitwieser, G.E.; Thakker, R.V. Large-scale exome datasets reveal a new class of adaptor-related protein complex 2 sigma subunit (AP2sigma) mutations, located at the interface with the AP2 alpha subunit, that impair calcium-sensing receptor signalling. Hum. Mol. Genet. 2018, 27, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Gorvin, C.M. Molecular and clinical insights from studies of calcium-sensing receptor mutations. J. Mol. Endocrinol. 2019, 63, R1–R16. [Google Scholar] [CrossRef] [Green Version]
- Pearce, S.H.; Wooding, C.; Davies, M.; Tollefsen, S.E.; Whyte, M.P.; Thakker, R.V. Calcium-sensing receptor mutations in familial hypocalciuric hypercalcaemia with recurrent pancreatitis. Clin. Endocrinol. 1996, 45, 675–680. [Google Scholar] [CrossRef]
- Sagi, S.V.; Joshi, H.; Trotman, J.; Elsey, T.; Swamy, A.; Rajkanna, J.; Bhat, N.A.; Haddadin, F.J.S.; Oyibo, S.O.; Park, S.-M. A novel CASR variant in a family with familial hypocalciuric hypercalcaemia and primary hyperparathyroidism. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020. [Google Scholar] [CrossRef]
- Eastell, R.; Brandi, M.L.; Costa, A.G.; D’Amour, P.; Shoback, D.M.; Thakker, R.V. Diagnosis of asymptomatic primary hyperparathyroidism: Proceedings of the Fourth International Workshop. J. Clin. Endocrinol. Metab. 2014, 99, 3570–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, S.J. Letter to the editor: Distinguishing typical primary hyperparathyroidism from familial hypocalciuric hypercalcemia by using an index of urinary calcium. J. Clin. Endocrinol. Metab. 2015, 100, L29–L30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, S.E.; Nissen, P.H.; Vestergaard, P.; Heickendorff, L.; Brixen, K.; Mosekilde, L. Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: A follow-up study on methods. Clin. Endocrinol. 2008, 69, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.F.; McAllister, J.; Merchant, A.; Rab, E.; Cook, J.; Eastell, R.; Balasubramanian, S. Urinary calcium indices in primary hyperparathyroidism (PHPT) and familial hypocalciuric hypercalcaemia (FHH): Which test performs best? Postgrad. Med. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Magno, A.L.; Leatherbarrow, K.M.; Brown, S.J.; Wilson, S.G.; Walsh, J.P.; Ward, B.K. Functional Analysis of Calcium-Sensing Receptor Variants Identified in Families Provisionally Diagnosed with Familial Hypocalciuric Hypercalcaemia. Calcif. Tissue Int. 2020, 107, 230–239. [Google Scholar] [CrossRef]
- Pidasheva, S.; Grant, M.; Canaff, L.; Ercan, O.; Kumar, U.; Hendy, G.N. Calcium-sensing receptor dimerizes in the endoplasmic reticulum: Biochemical and biophysical characterization of CASR mutants retained intracellularly. Hum. Mol. Genet. 2006, 15, 2200–2209. [Google Scholar] [CrossRef] [Green Version]
- Leach, K.; Wen, A.; Cook, A.E.; Sexton, P.M.; Conigrave, A.D.; Christopoulos, A. Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 2013, 154, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Gorvin, C.M.; Cranston, T.; Hannan, F.M.; Rust, N.; Qureshi, A.; Nesbit, M.A.; Thakker, R.V. A G-protein Subunit-α11 Loss-of-Function Mutation, Thr54Met, Causes Familial Hypocalciuric Hypercalcemia Type 2 (FHH2). J. Bone Miner. Res. 2016, 31, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Nesbit, M.A.; Hannan, F.M.; Howles, S.A.; Babinsky, V.N.; Head, R.A.; Cranston, T.; Rust, N.; Hobbs, M.R.; Heath, H., III; Thakker, R.V. Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. N. Engl. J. Med. 2013, 368, 2476–2486. [Google Scholar] [CrossRef] [Green Version]
- Gorvin, C.M.; Hannan, F.M.; Cranston, T.; Valta, H.; Makitie, O.; Schalin-Jantti, C.; Thakker, R.V. Cinacalcet Rectifies Hypercalcemia in a Patient with Familial Hypocalciuric Hypercalcemia Type 2 (FHH2) Caused by a Germline Loss-of-Function Galpha11 Mutation. J. Bone Miner. Res. 2018, 33, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Gorvin, C.M.; Rogers, A.; Hastoy, B.; Tarasov, A.I.; Frost, M.; Sposini, S.; Inoue, A.; Whyte, M.P.; Rorsman, P.; Hanyaloglu, A.C.; et al. AP2sigma Mutations Impair Calcium-Sensing Receptor Trafficking and Signaling, and Show an Endosomal Pathway to Spatially Direct G-Protein Selectivity. Cell Rep. 2018, 22, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M.R.; Chou, Y.H.; Marx, S.J.; Steinmann, B.; Cole, D.E.; Brandi, M.L.; Papapoulos, S.E.; Menko, F.H.; Hendy, G.N.; Brown, E.M.; et al. Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J. Clin. Investig. 1994, 93, 1108–1112. [Google Scholar] [CrossRef] [Green Version]
- Hannan, F.M.; Nesbit, M.A.; Christie, P.T.; Lissens, W.; van der Schueren, B.; Bex, M.; Bouillon, R.; Thakker, R.V. A homozygous inactivating calcium-sensing receptor mutation, Pro339Thr, is associated with isolated primary hyperparathyroidism: Correlation between location of mutations and severity of hypercalcaemia. Clin. Endocrinol. 2010, 73, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Marx, S.J.; Sinaii, N. Neonatal Severe Hyperparathyroidism: Novel Insights from Calcium, PTH, and the CASR Gene. J. Clin. Endocrinol. Metab. 2020, 105, 1061–1078. [Google Scholar] [CrossRef]
- Lee, J.Y.; Shoback, D.M. Familial hypocalciuric hypercalcemia and related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 609–619. [Google Scholar] [CrossRef]
- Gannon, A.W.; Monk, H.M.; Levine, M.A. Cinacalcet monotherapy in neonatal severe hyperparathyroidism: A case study and review. J. Clin. Endocrinol. Metab. 2014, 99, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Mayr, B.M.; Schnabel, D.; Dörr, H.-G.; Schöfl, C. Genetics in Endocrinology: Gain and loss of function mutations of the calcium-sensing receptor and associated proteins: Current treatment concepts. Eur. J. Endocrinol. 2016, 174, R189–R208. [Google Scholar] [CrossRef] [Green Version]
- Pearce, S.H.; Williamson, C.; Kifor, O.; Bai, M.; Coulthard, M.G.; Davies, M.; Lewis-Barned, N.; McCredie, D.; Powell, H.; Kendall-Taylor, P.; et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N. Engl. J. Med. 1996, 335, 1115–1122. [Google Scholar] [CrossRef]
- Roszko, K.L.; Bi, R.D.; Mannstadt, M. Autosomal Dominant Hypocalcemia (Hypoparathyroidism) Types 1 and 2. Front. Physiol. 2016, 7, 458. [Google Scholar] [CrossRef] [Green Version]
- Dershem, R.; Gorvin, C.M.; Metpally, R.P.; Krishnamurthy, S.; Smelser, D.T.; Hannan, F.M.; Carey, D.J.; Thakker, R.V.; Breitwieser, G.E. Familial Hypocalciuric Hypercalcemia Type 1 and Autosomal-Dominant Hypocalcemia Type 1: Prevalence in a Large Healthcare Population. Am. J. Hum. Genet. 2020, 106, 734–747. [Google Scholar] [CrossRef]
- Hannan, F.M.; Thakker, R.V. Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 359–371. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Gole, E.; Melachroinou, K.; Trangas, T.; Bountouvi, E.; Papadimitriou, A. Clinical characterization of a novel calcium sensing receptor genetic alteration in a Greek patient with autosomal dominant hypocalcemia type 1. Hormones 2017, 16, 200–204. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Fukumoto, S.; Chang, H.; Takeuchi, Y.; Hasegawa, Y.; Okazaki, R.; Chikatsu, N.; Fujita, T. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet 2002, 360, 692–694. [Google Scholar] [CrossRef]
- Hu, J.; Spiegel, A.M. Structure and function of the human calcium-sensing receptor: Insights from natural and engineered mutations and allosteric modulators. J. Cell. Mol. Med. 2007, 11, 908–922. [Google Scholar] [CrossRef]
- Dore, A.S.; Okrasa, K.; Patel, J.C.; Serrano-Vega, M.; Bennett, K.; Cooke, R.M.; Errey, J.C.; Jazayeri, A.; Khan, S.; Tehan, B.; et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 2014, 511, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Leach, K.; Wen, A.; Davey, A.E.; Sexton, P.M.; Conigrave, A.D.; Christopoulos, A. Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology 2012, 153, 4304–4316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Mulpuri, N.; Hannan, F.M.; Nesbit, M.A.; Thakker, R.V.; Hamelberg, D.; Brown, E.M.; Yang, J.J. Role of Ca2+ and L-Phe in regulating functional cooperativity of disease-associated “toggle” calcium-sensing receptor mutations. PLoS ONE 2014, 9, e113622. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Opas, E.E.; Tuluc, F.; Metzger, D.L.; Hou, C.; Hakonarson, H.; Levine, M.A. Autosomal dominant hypoparathyroidism caused by germline mutation in GNA11: Phenotypic and molecular characterization. J. Clin. Endocrinol. Metab. 2014, 99, E1774–E1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piret, S.E.; Gorvin, C.M.; Pagnamenta, A.T.; Howles, S.A.; Cranston, T.; Rust, N.; Nesbit, M.A.; Glaser, B.; Taylor, J.C.; Buchs, A.E.; et al. Identification of a G-Protein Subunit-alpha11 Gain-of-Function Mutation, Val340Met, in a Family with Autosomal Dominant Hypocalcemia Type 2 (ADH2). J. Bone Miner. Res. 2016, 31, 1207–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Hasegawa, Y.; Nakae, J.; Nanao, K.; Takahashi, I.; Tajima, T.; Shinohara, N.; Fujieda, K. Hydrochlorothiazide effectively reduces urinary calcium excretion in two Japanese patients with gain-of-function mutations of the calcium-sensing receptor gene. J. Clin. Endocrinol. Metab. 2002, 87, 3068–3073. [Google Scholar] [CrossRef]
- Dong, B.; Endo, I.; Ohnishi, Y.; Kondo, T.; Hasegawa, T.; Amizuka, N.; Kiyonari, H.; Shioi, G.; Abe, M.; Fukumoto, S.; et al. Calcilytic Ameliorates Abnormalities of Mutant Calcium-Sensing Receptor (CaSR) Knock-In Mice Mimicking Autosomal Dominant Hypocalcemia (ADH). J. Bone Miner. Res. 2015, 30, 1980–1993. [Google Scholar] [CrossRef]
- Cinque, L.; Sparaneo, A.; Penta, L.; Mencarelli, A.; Rogaia, D.; Esposito, S.; Fabrizio, F.P.; Baorda, F.; Verrotti, A.; Falorni, A.; et al. Autosomal Dominant PTH Gene Signal Sequence Mutation in a Family with Familial Isolated Hypoparathyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 3961–3969. [Google Scholar] [CrossRef]
- Hendy, G.N.; Cole, D.E.C. Familial isolated hypoparathyroidism. In Hypoparathyroidism; Brandi, M.L., Brown, E., Eds.; Springer: Milan, Italy, 2015. [Google Scholar]
- Garcia-Castano, A.; Madariaga, L.; Gomez-Conde, S.; Cordo, C.L.R.; Lopez-Iglesias, M.; Garcia-Fernandez, Y.; Martin, A.; Gonzalez, P.; Goicolea, I.; de Nanclares, G.P.; et al. Five patients with disorders of calcium metabolism presented with GCM2 gene variants. Sci. Rep. 2021, 11, 2968. [Google Scholar] [CrossRef]
- Hendy, G.N.; Cole, D.E.C.; Bastepe, M. Hypoparathyroidism and Pseudohypoparathyroidism. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Schipani, E.; Jensen, G.S.; Pincus, J.; Nissenson, R.A.; Gardella, T.J.; Juppner, H. Constitutive activation of the cyclic adenosine 3′,5′-monophosphate signaling pathway by parathyroid hormone (PTH)/PTH-related peptide receptors mutated at the two loci for Jansen’s metaphyseal chondrodysplasia. Mol. Endocrinol. 1997, 11, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Bastepe, M. The GNAS locus and pseudohypoparathyroidism. Adv. Exp. Med. Biol. 2008, 626, 27–40. [Google Scholar] [CrossRef]
- Mantovani, G.; Bastepe, M.; Monk, D.; de Sanctis, L.; Thiele, S.; Usardi, A.; Ahmed, S.F.; Bufo, R.; Choplin, T.; de Filippo, G.; et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: First international Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 476–500. [Google Scholar] [CrossRef]
- De Nanclares, G.P.; Fernandez-Rebollo, E.; Santin, I.; Garcia-Cuartero, B.; Gaztambide, S.; Menendez, E.; Morales, M.J.; Pombo, M.; Bilbao, J.R.; Barros, F.; et al. Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright’s hereditary osteodystrophy. J. Clin. Endocrinol. Metab. 2007, 92, 2370–2373. [Google Scholar] [CrossRef] [Green Version]
- Albright, F.; Forbes, A.P.; Henneman, P.H. Pseudo-pseudohypoparathyroidism. Trans. Assoc. Am. Physicians 1952, 65, 337–350. [Google Scholar]
- Plagge, A.; Kelsey, G.; Germain-Lee, E.L. Physiological functions of the imprinted Gnas locus and its protein variants Galpha(s) and XLalpha(s) in human and mouse. J. Endocrinol. 2008, 196, 193–214. [Google Scholar] [CrossRef] [Green Version]
- Thiele, S.; de Sanctis, L.; Werner, R.; Grotzinger, J.; Aydin, C.; Juppner, H.; Bastepe, M.; Hiort, O. Functional characterization of GNAS mutations found in patients with pseudohypoparathyroidism type Ic defines a new subgroup of pseudohypoparathyroidism affecting selectively Gsalpha-receptor interaction. Hum. Mutat. 2011, 32, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Litman, D.; Rosenberg, M.J.; Yu, S.; Biesecker, L.G.; Weinstein, L.S. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J. Clin. Investig. 2000, 106, 1167–1174. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Nealon, J.G.; Weinstein, L.S. Distinct patterns of abnormal GNAS imprinting in familial and sporadic pseudohypoparathyroidism type IB. Hum. Mol. Genet. 2005, 14, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Bastepe, M.; Frohlich, L.F.; Hendy, G.N.; Indridason, O.S.; Josse, R.G.; Koshiyama, H.; Korkko, J.; Nakamoto, J.M.; Rosenbloom, A.L.; Slyper, A.H.; et al. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J. Clin. Investig. 2003, 112, 1255–1263. [Google Scholar] [CrossRef]
- Drezner, M.; Neelon, F.A.; Lebovitz, H.E. Pseudohypoparathyroidism type II: A possible defect in the reception of the cyclic AMP signal. N. Engl. J. Med. 1973, 289, 1056–1060. [Google Scholar] [CrossRef]
- Miller, W.L. Genetic disorders of Vitamin D biosynthesis and degradation. J. Steroid Biochem. Mol. Biol. 2017, 165, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Nicolaidou, P.; Tsitsika, A.; Papadimitriou, A.; Karantana, A.; Papadopoulou, A.; Psychou, F.; Liakopoulou, D.; Georgouli, H.; Kakourou, T.; Chrousos, G. Hereditary Vitamin D-resistant Rickets in Greek Children: Genotype, Phenotype, and Long-term Response to Treatment. J. Pediatr. Endocrinol. Metab. 2007, 20, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Malloy, P.J.; Wang, J.; Srivastava, T.; Feldman, D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia resulting from a novel missense mutation in the DNA-binding domain of the vitamin D receptor. Mol. Genet. Metab. 2010, 99, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaidou, P.; Papadopoulou, A.; Georgouli, H.; Matsinos, Y.; Tsapra, H.; Fretzayas, A.; Giannoulia-Karantana, A.; Kitsiou, S.; Douros, K.; Papassotiriou, I.; et al. Calcium and Vitamin D Metabolism in Hypocalcemic Vitamin D-Resistant Rickets Carriers. Horm. Res. Paediatr. 2006, 65, 83–88. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Bountouvi, E.; Gole, E.; Doulgeraki, A.; Tournis, S.; Papadimitriou, A.; Nicolaidou, P. Identification of a novel nonsense mutation in the ligand-binding domain of the vitamin d receptor gene and clinical description of two greek patients with hereditary vitamin d-resistant rickets and alopecia. Horm. Res. Paediatr. 2014, 82, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Akinci, A.; Dundar, I.; Kivilcim, M. The Effectiveness of Cinacalcet as an Adjunctive Therapy for Hereditary 1,25 Dihydroxyvitamin D3-Resistant Rickets. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 172–178. [Google Scholar] [CrossRef]
- Lucas, J.; Badia, J.L.; Lucas, E.; Remon, A. Cinacalcet treatment experience in hereditary vitamin D resistant rickets. J. Pediatr. Endocrinol. Metab. 2020, 33, 313–318. [Google Scholar] [CrossRef]
- Hochberg, Z.; Benderli, A.; Levy, J.; Vardi, P.; Weisman, Y.; Chen, T.; Feldman, D. 1,25-Dihydroxyvitamin D resistance, rickets, and alopecia. Am. J. Med. 1984, 77, 805–811. [Google Scholar] [CrossRef]
- Xu, H.; Bai, L.; Collins, J.F.; Ghishan, F.K. Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1,25-(OH)(2) vitamin D(3). Am. J. Physiol. Cell Physiol. 2002, 282, C487–C493. [Google Scholar] [CrossRef] [Green Version]
- Biber, J.; Hernando, N.; Forster, I. Phosphate transporters and their function. Annu. Rev. Physiol. 2013, 75, 535–550. [Google Scholar] [CrossRef]
- Goyal, R.; Jialal, I. Hyperphosphatemia; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Blaine, J.; Okamura, K.; Giral, H.; Breusegem, S.; Caldas, Y.; Millard, A.; Barry, N.; Levi, M. PTH-induced internalization of apical membrane NaPi2a: Role of actin and myosin VI. Am. J. Physiol. Cell Physiol. 2009, 297, C1339–C1346. [Google Scholar] [CrossRef] [Green Version]
- Centeno, P.; Herberger, A.; Mun, H.-C.; Tu, C.; Nemeth, E.F.; Chang, W.; Conigrave, A.D.; Ward, D.T. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat Commun. 2019, 10, 4693. [Google Scholar] [CrossRef]
- Katai, K.; Miyamoto, K.; Kishida, S.; Segawa, H.; Nii, T.; Tanaka, H.; Tani, Y.; Arai, H.; Tatsumi, S.; Morita, K.; et al. Regulation of intestinal Na+-dependent phosphate co-transporters by a low-phosphate diet and 1,25-dihydroxyvitamin D3. Biochem. J. 1999, 343, 705–712. [Google Scholar] [CrossRef]
- Bacchetta, J.; Bardet, C.; Prie, D. Physiology of FGF23 and overview of genetic diseases associated with renal phosphate wasting. Metabolism 2020, 103, 153865. [Google Scholar] [CrossRef]
- Kaludjerovic, J.; Komaba, H.; Sato, T.; Erben, R.G.; Baron, R.; Olauson, H.; Larsson, T.E.; Lanske, B. Klotho expression in long bones regulates FGF23 production during renal failure. FASEB J. 2017, 31, 2050–2064. [Google Scholar] [CrossRef] [Green Version]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- Sprague, S.M.; Wetmore, J.B.; Gurevich, K.; Da Roza, G.; Buerkert, J.; Reiner, M.; Goodman, W.; Cooper, K. Effect of Cinacalcet and Vitamin D Analogs on Fibroblast Growth Factor-23 during the Treatment of Secondary Hyperparathyroidism. Clin. J. Am. Soc. Nephrol. 2015, 10, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2015, 34, 132–139. [Google Scholar] [CrossRef]
- Gattineni, J.; Bates, C.; Twombley, K.; Dwarakanath, V.; Robinson, M.L.; Goetz, R.; Mohammadi, M.; Baum, M. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am. J. Physiol. Renal. Physiol. 2009, 297, F282–F291. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, Y.; Fukumoto, S. X-Linked Hypophosphatemia and FGF23-Related Hypophosphatemic Diseases: Prospect for New Treatment. Endocr. Rev. 2018, 39, 274–291. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.; Durlacher, K.; Silver, J.; Naveh-Many, T.; Levi, R. The fibroblast growth factor receptor mediates the increased FGF23 expression in acute and chronic uremia. Am. J. Physiol. Renal. Physiol. 2016, 310, F217–F221. [Google Scholar] [CrossRef] [Green Version]
- Wohrle, S.; Bonny, O.; Beluch, N.; Gaulis, S.; Stamm, C.; Scheibler, M.; Muller, M.; Kinzel, B.; Thuery, A.; Brueggen, J.; et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J. Bone Miner. Res. 2011, 26, 2486–2497. [Google Scholar] [CrossRef]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor Specificity of the Fibroblast Growth Factor Family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [Green Version]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-O, M.; Nabeshima, Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef]
- Andrukhova, O.; Bayer, J.; Schuler, C.; Zeitz, U.; Murali, S.K.; Ada, S.; Alvarez-Pez, J.M.; Smorodchenko, A.; Erben, R.G. Klotho Lacks an FGF23-Independent Role in Mineral Homeostasis. J. Bone Miner. Res. 2017, 32, 2049–2061. [Google Scholar] [CrossRef] [Green Version]
- Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta 2009, 1790, 1049–1058. [Google Scholar] [CrossRef]
- Wagner, C.A.; Rubio-Aliaga, I.; Hernando, N. Renal phosphate handling and inherited disorders of phosphate reabsorption: An update. Pediatr. Nephrol. 2019, 34, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Tieder, M.; Modai, D.; Samuel, R.; Arie, R.; Halabe, A.; Bab, I.; Gabizon, D.; Liberman, U.A. Hereditary Hypophosphatemic Rickets with Hypercalciuria. N. Engl. J. Med. 1985, 312, 611–617. [Google Scholar] [CrossRef]
- Bergwitz, C.; Roslin, N.M.; Tieder, M.; Loredo-Osti, J.C.; Bastepe, M.; Abu-Zahra, H.; Frappier, D.; Burkett, K.; Carpenter, T.O.; Anderson, D.; et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am. J. Hum. Genet. 2006, 78, 179–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz-Depiereux, B.; Benet-Pages, A.; Eckstein, G.; Tenenbaum-Rakover, Y.; Wagenstaller, J.; Tiosano, D.; Gershoni-Baruch, R.; Albers, N.; Lichtner, P.; Schnabel, D.; et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am. J. Hum. Genet. 2006, 78, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergwitz, C.; Miyamoto, K.I. Hereditary hypophosphatemic rickets with hypercalciuria: Pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch. 2019, 471, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Ro, H.; Mundra, V.R.R.; Joseph, K.; Brenner, D.; Carpenter, T.O.; Rizk, D.V.; Bergwitz, C. Description of 5 Novel SLC34A3/NPT2c Mutations Causing Hereditary Hypophosphatemic Rickets with Hypercalciuria. Kidney Int. Rep. 2019, 4, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, S.; Imel, E.A.; Kreiter, M.L.; Yu, X.; Mackenzie, D.S.; Sorenson, A.H.; Goetz, R.; Mohammadi, M.; White, K.E.; Econs, M.J. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J. Musculoskelet Neuronal Interact 2007, 7, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, D.; Wee, M.J.; Reyes, M.; Li, Y.; Simm, P.J.; Sharma, A.; Schlingmann, K.P.; Janner, M.; Biggin, A.; Lazier, J.; et al. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J. Am. Soc. Nephrol. 2014, 25, 2366–2375. [Google Scholar] [CrossRef] [Green Version]
- Kremke, B.; Bergwitz, C.; Ahrens, W.; Schutt, S.; Schumacher, M.; Wagner, V.; Holterhus, P.M.; Juppner, H.; Hiort, O. Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/NaPi-IIc can be masked by vitamin D deficiency and can be associated with renal calcifications. Exp. Clin. Endocrinol. Diabetes 2009, 117, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Yavropoulou, M.P.; Kotsa, K.; Gotzamani Psarrakou, A.; Papazisi, A.; Tranga, T.; Ventis, S.; Yovos, J.G. Cinacalcet in hyperparathyroidism secondary to X-linked hypophosphatemic rickets: Case report and brief literature review. Hormones 2010, 9, 274–278. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, T.O.; Imel, E.A.; Holm, I.A.; Jan de Beur, S.M.; Insogna, K.L. A clinician’s guide to X-linked hypophosphatemia. J. Bone Miner. Res. 2011, 26, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Ruchon, A.F.; Tenenhouse, H.S.; Marcinkiewicz, M.; Siegfried, G.; Aubin, J.E.; DesGroseillers, L.; Crine, P.; Boileau, G. Developmental Expression and Tissue Distribution of Phex Protein: Effect of the Hyp Mutation and Relationship to Bone Markers. J. Bone Miner. Res. 2000, 15, 1440–1450. [Google Scholar] [CrossRef]
- Srivastava, T.; Alon, U.S. Cinacalcet as adjunctive therapy for hereditary 1,25-dihydroxyvitamin D-resistant rickets. J. Bone Miner. Res. 2013, 28, 992–996. [Google Scholar] [CrossRef]
- Alon, U.S.; Jarka, D.; Monachino, P.J.; VanSickle, J.S.; Srivastava, T. Cinacalcet as an alternative to phosphate therapy in X-linked hypophosphataemic rickets. Clin. Endocrinol. 2017, 87, 114–116. [Google Scholar] [CrossRef]
- Lamb, Y.N. Burosumab: First Global Approval. Drugs 2018, 78, 707–714. [Google Scholar] [CrossRef]
- Florenzano, P.; Hartley, I.R.; Jimenez, M.; Roszko, K.; Gafni, R.I.; Collins, M.T. Tumor-Induced Osteomalacia. Calcif. Tissue Int. 2021, 108, 128–142. [Google Scholar] [CrossRef]
- Imel, E.A.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Ward, L.M.; Nilsson, O.; Simmons, J.H.; Padidela, R.; Namba, N.; Cheong, H.I.; et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: A randomised, active-controlled, open-label, phase 3 trial. Lancet 2019, 393, 2416–2427. [Google Scholar] [CrossRef]
- Shimada, T.; Muto, T.; Urakawa, I.; Yoneya, T.; Yamazaki, Y.; Okawa, K.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 2002, 143, 3179–3182. [Google Scholar] [CrossRef]
- Gattineni, J.; Baum, M. Genetic disorders of phosphate regulation. Pediatr. Nephrol. 2012, 27, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Gohil, A.; Imel, E.A. FGF23 and Associated Disorders of Phosphate Wasting. Pediatr. Endocrinol. Rev. 2019, 17, 17–34. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, Z.; Wang, O.; Li, M.; Xing, X.; Hsieh, E.; Fukumoto, S.; Jiang, Y.; Xia, W. Earlier Onset in Autosomal Dominant Hypophosphatemic Rickets of R179 than R176 Mutations in Fibroblast Growth Factor 23: Report of 20 Chinese Cases and Review of the Literature. Calcif. Tissue Int. 2019, 105, 476–486. [Google Scholar] [CrossRef]
- Imel, E.A.; Hui, S.L.; Econs, M.J. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J. Bone Miner. Res. 2007, 22, 520–526. [Google Scholar] [CrossRef]
- Imel, E.A.; Peacock, M.; Gray, A.K.; Padgett, L.R.; Hui, S.L.; Econs, M.J. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J. Clin. Endocrinol. Metab. 2011, 96, 3541–3549. [Google Scholar] [CrossRef]
- Imel, E.A.; Liu, Z.; Coffman, M.; Acton, D.; Mehta, R.; Econs, M.J. Oral Iron Replacement Normalizes Fibroblast Growth Factor 23 in Iron-Deficient Patients with Autosomal Dominant Hypophosphatemic Rickets. J. Bone Miner. Res. 2020, 35, 231–238. [Google Scholar] [CrossRef]
- Athonvarangkul, D.; Insogna, K.L. New Therapies for Hypophosphatemia-Related to FGF23 Excess. Calcif. Tissue Int. 2021, 108, 143–157. [Google Scholar] [CrossRef]
- Kuthiroly, S.; Yesodharan, D.; Ghosh, A.; White, K.E.; Nampoothiri, S. Osteoglophonic Dysplasia: Phenotypic and Radiological Clues. J. Pediatr. Genet. 2017, 6, 247–251. [Google Scholar] [CrossRef]
- Courage, C.; Jackson, C.B.; Owczarek-Lipska, M.; Jamsheer, A.; Sowinska-Seidler, A.; Piotrowicz, M.; Jakubowski, L.; Dalleves, F.; Riesch, E.; Neidhardt, J.; et al. Novel synonymous and missense variants in FGFR1 causing Hartsfield syndrome. Am. J. Med. Genet. A 2019, 179, 2447–2453. [Google Scholar] [CrossRef] [PubMed]
- Lorenz-Depiereux, B.; Bastepe, M.; Benet-Pagès, A.; Amyere, M.; Wagenstaller, J.; Müller-Barth, U.; Badenhoop, K.; Kaiser, S.M.; Rittmaster, R.S.; Shlossberg, A.H.; et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat. Genet. 2006, 38, 1248–1250. [Google Scholar] [CrossRef]
- Levy-Litan, V.; Hershkovitz, E.; Avizov, L.; Leventhal, N.; Bercovich, D.; Chalifa-Caspi, V.; Manor, E.; Buriakovsky, S.; Hadad, Y.; Goding, J.; et al. Autosomal-Recessive Hypophosphatemic Rickets is Associated with an Inactivation Mutation in the ENPP1 Gene. Am. J. Hum. Genet. 2010, 86, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Negri, A.L. Hereditary hypophosphatemias: New genes in the bone-kidney axis. Nephrology 2007, 12, 317–320. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, B.; Qin, C.; Cao, Z.; Xie, Y.; Dallas, S.L.; McKee, M.D.; Drezner, M.K.; Bonewald, L.F.; Feng, J.Q. The biological function of DMP-1 in osteocyte maturation is mediated by its 57-kDa C-terminal fragment. J. Bone Miner. Res. 2011, 26, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Li, X.; Zhang, Q.; Liu, C.; Gong, Y.; Wang, O.; Li, M.; Xing, X.; Jiang, Y.; Xia, W. Clinical Characteristics and Bone Features of Autosomal Recessive Hypophosphatemic Rickets Type 1 in Three Chinese Families: Report of Five Chinese Cases and Review of the Literature. Calcif. Tissue Int. 2020, 107, 636–648. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Höhne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with ’idiopathic’ infantile arterial calcification. Nat. Genet. 2003, 34, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Kotwal, A.; Ferrer, A.; Kumar, R.; Singh, R.J.; Murthy, V.; Schultz-Rogers, L.; Zimmermann, M.; Lanpher, B.; Zimmerman, K.; Stabach, P.R.; et al. Clinical and Biochemical Phenotypes in a Family with ENPP1 Mutations. J. Bone Miner. Res. 2020, 35, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Rafaelsen, S.H.; Raeder, H.; Fagerheim, A.K.; Knappskog, P.; Carpenter, T.O.; Johansson, S.; Bjerknes, R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J. Bone Miner. Res. 2013, 28, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Appaiah, H.N.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef] [Green Version]
- Brownstein, C.A.; Adler, F.; Nelson-Williams, C.; Iijima, J.; Li, P.; Imura, A.; Nabeshima, Y.; Reyes-Mugica, M.; Carpenter, T.O.; Lifton, R.P. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc. Natl. Acad. Sci. USA 2008, 105, 3455–3460. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.A.; Rubio-Aliaga, I.; Biber, J.; Hernando, N. Genetic diseases of renal phosphate handling. Nephrol. Dial. Transpl. 2014, 29 (Suppl. 4), iv45–iv54. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulou, A.; Bountouvi, E.; Karachaliou, F.-E. The Molecular Basis of Calcium and Phosphorus Inherited Metabolic Disorders. Genes 2021, 12, 734. https://doi.org/10.3390/genes12050734
Papadopoulou A, Bountouvi E, Karachaliou F-E. The Molecular Basis of Calcium and Phosphorus Inherited Metabolic Disorders. Genes. 2021; 12(5):734. https://doi.org/10.3390/genes12050734
Chicago/Turabian StylePapadopoulou, Anna, Evangelia Bountouvi, and Fotini-Eleni Karachaliou. 2021. "The Molecular Basis of Calcium and Phosphorus Inherited Metabolic Disorders" Genes 12, no. 5: 734. https://doi.org/10.3390/genes12050734
APA StylePapadopoulou, A., Bountouvi, E., & Karachaliou, F.-E. (2021). The Molecular Basis of Calcium and Phosphorus Inherited Metabolic Disorders. Genes, 12(5), 734. https://doi.org/10.3390/genes12050734