
Signatures of Transcription Factor Evolution and the Secondary Gain of Red Algae Complexity

 , , , and
, , , and 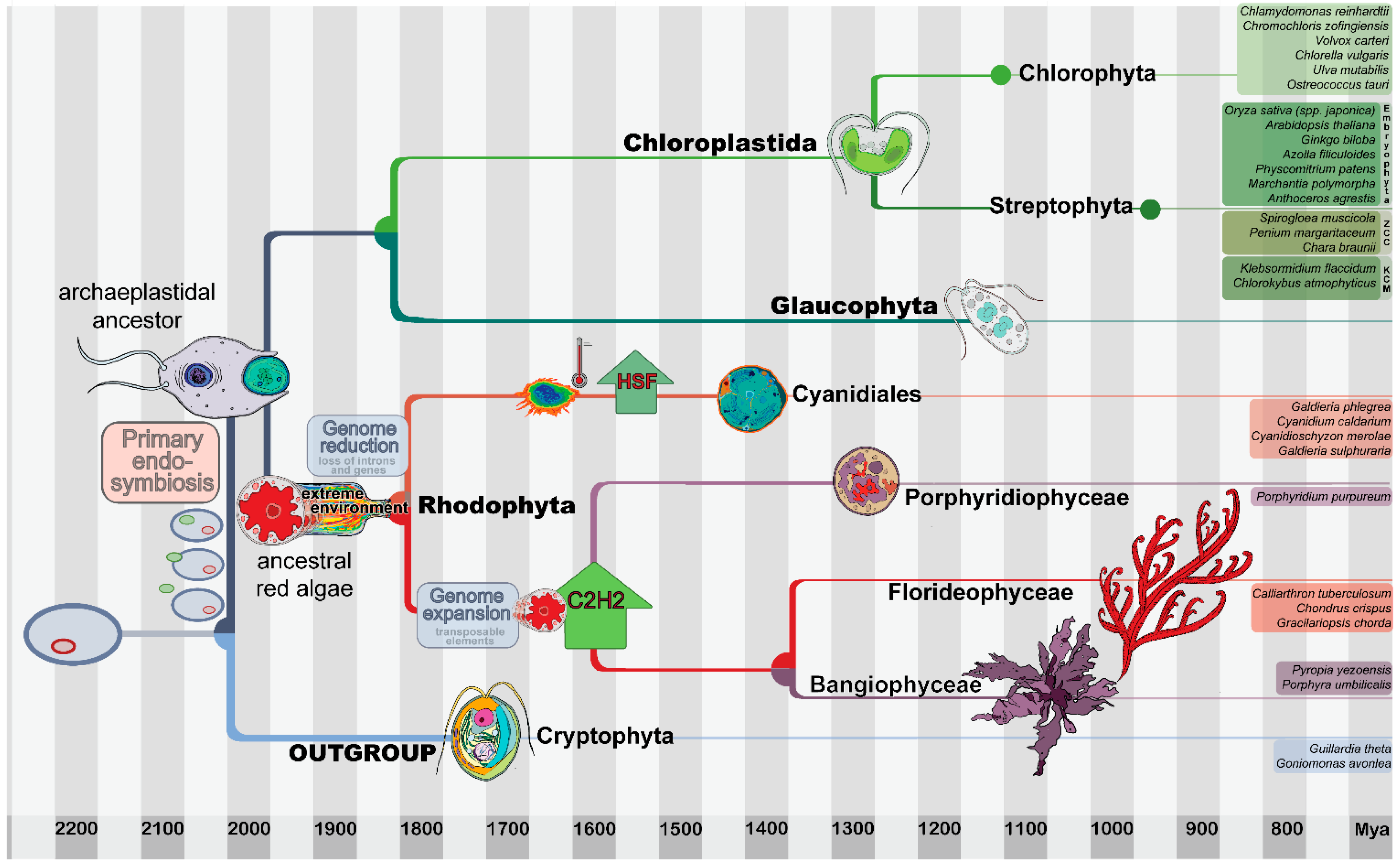{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Materials and Methods
2.1. Dataset
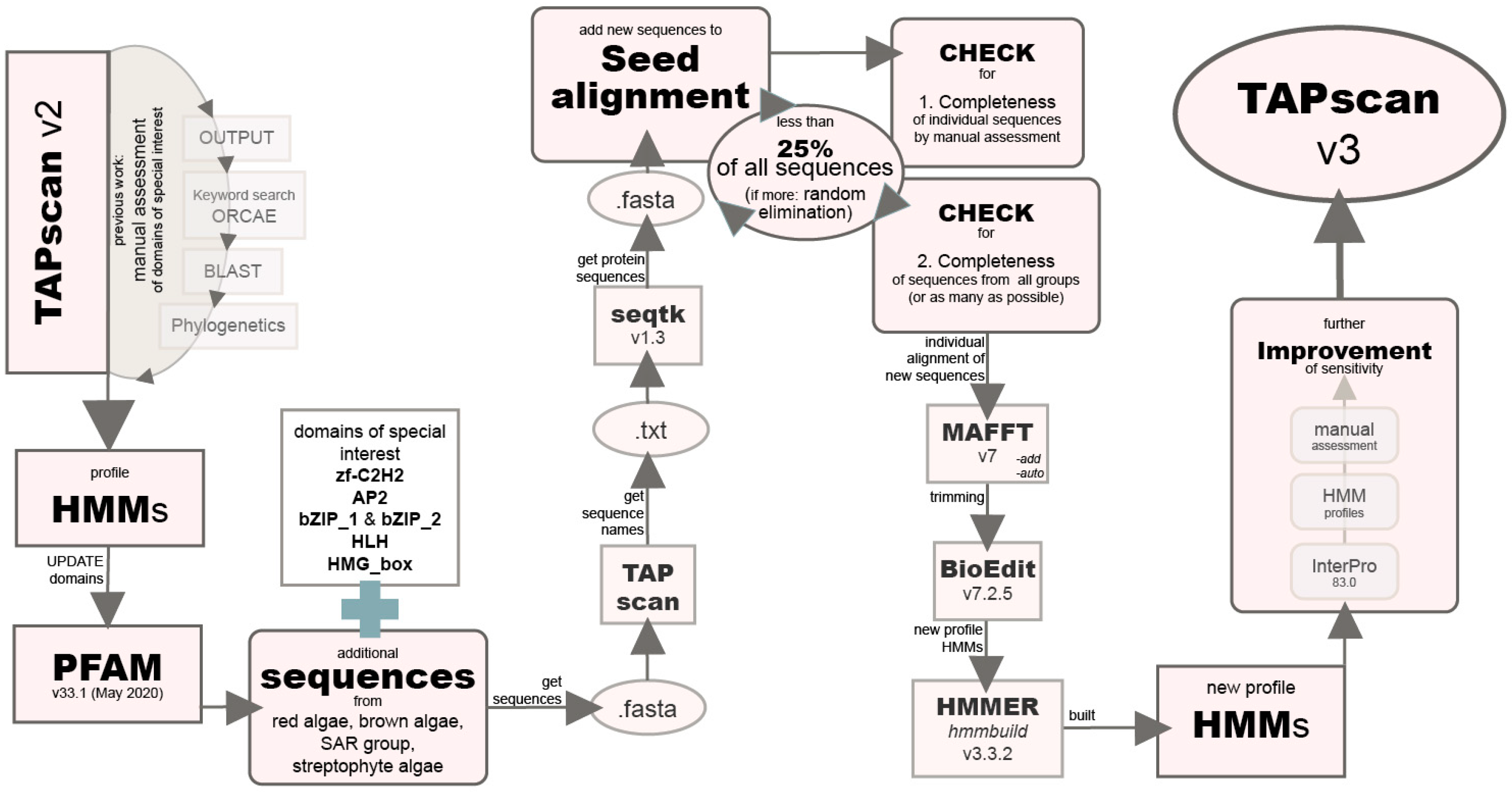
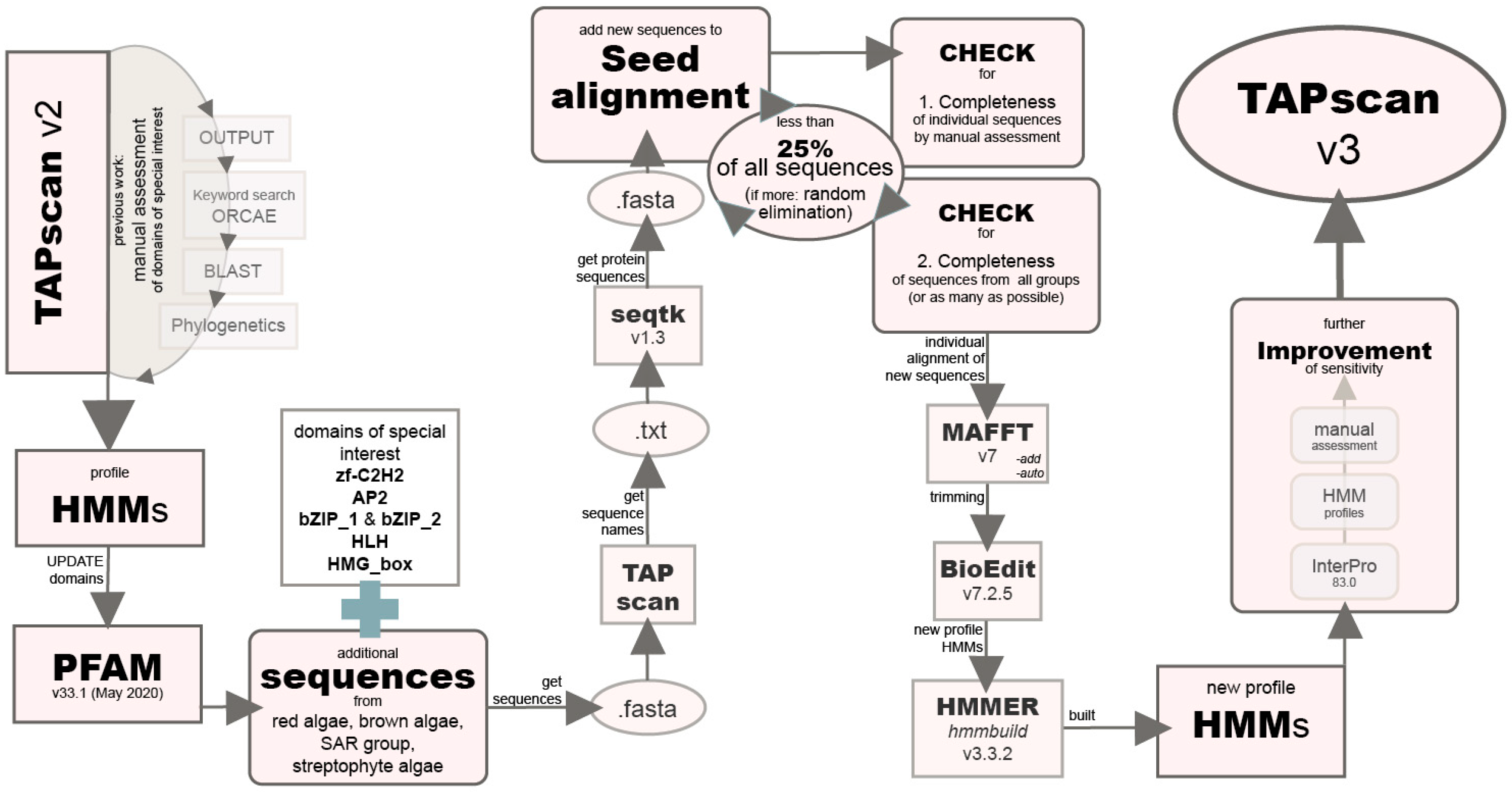
2.2. Improving the Sensitivity of Selected Profile HMMs for Algae by Adding Diverse Algal Sequences
2.3. New TAPscan Version
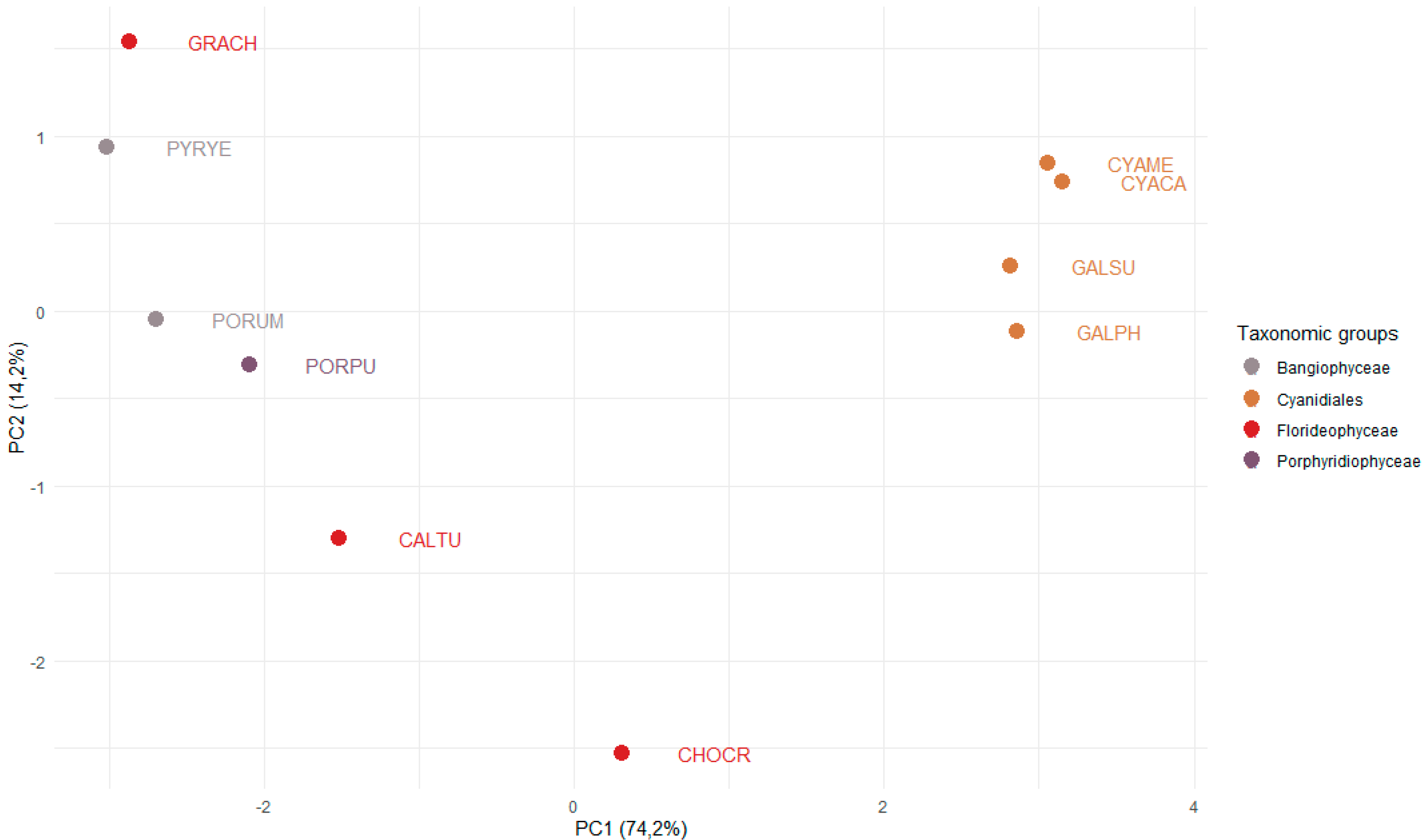
2.4. Comparisons of Unicellular and Multicellular Red Algae
2.5. Calculation of Expansions/Contractions and Gains/Losses
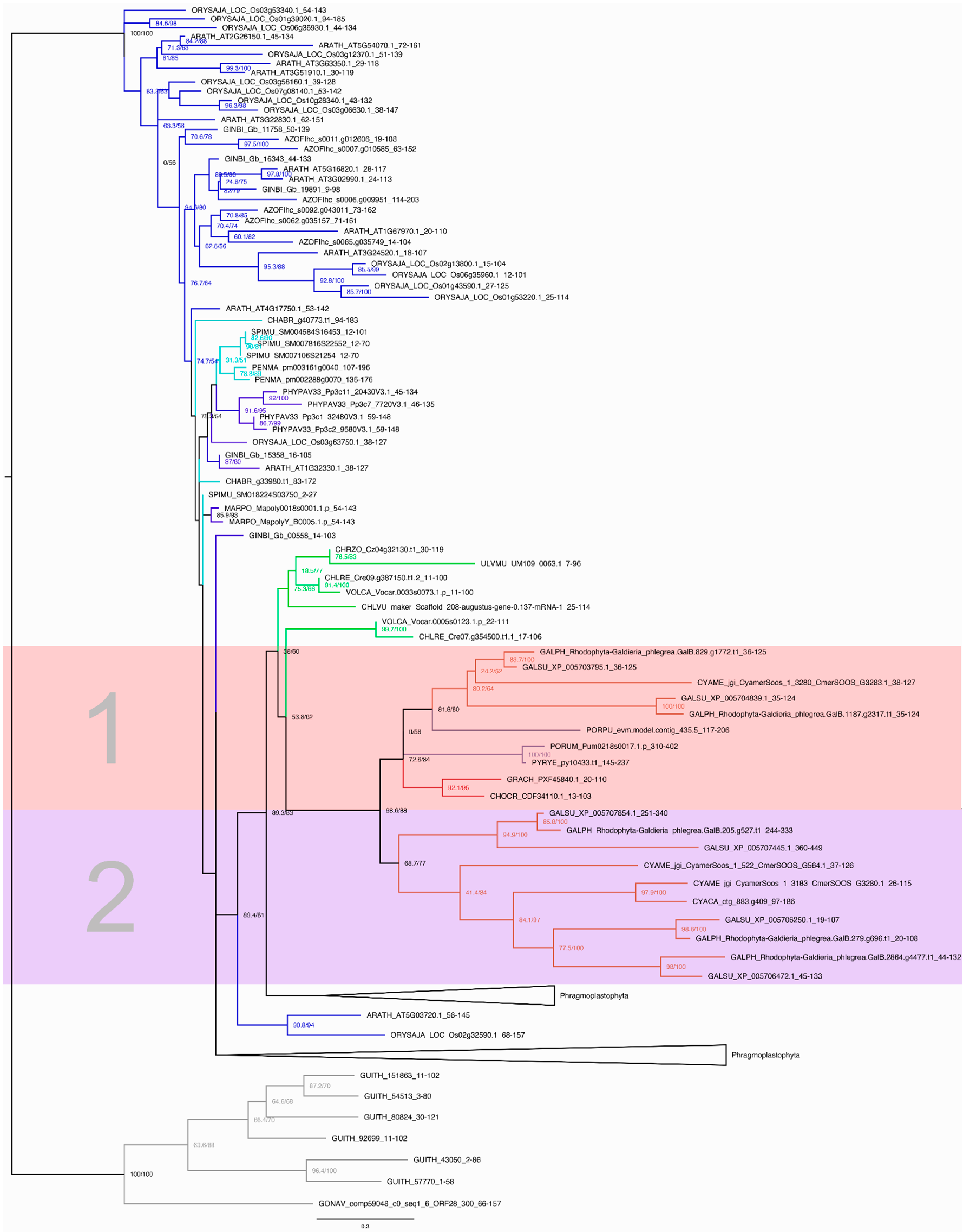
2.6. Phylogenetic Inference
3. Results and Discussion
3.1. Improved Sensitivity for the Detection of Five TAP Families
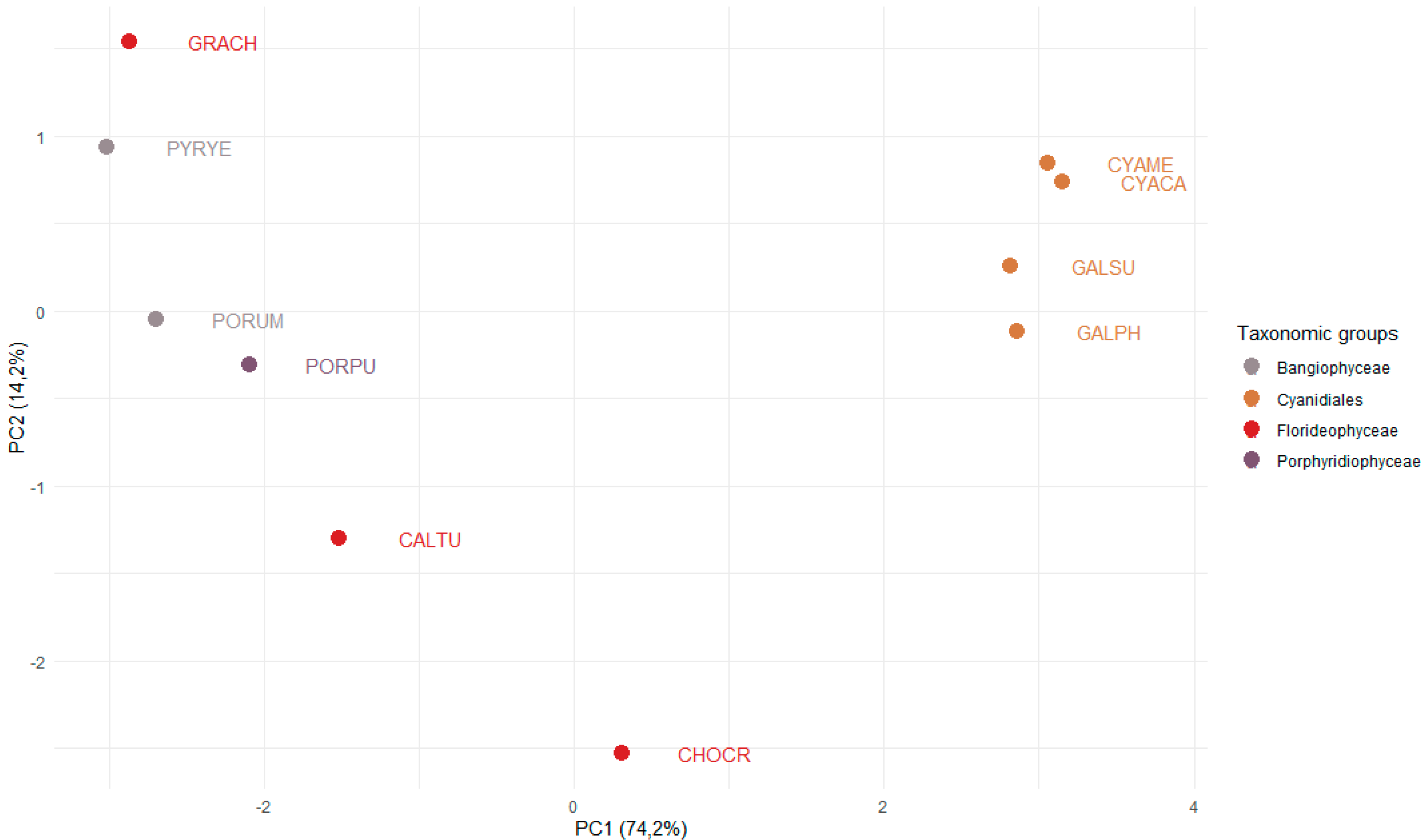
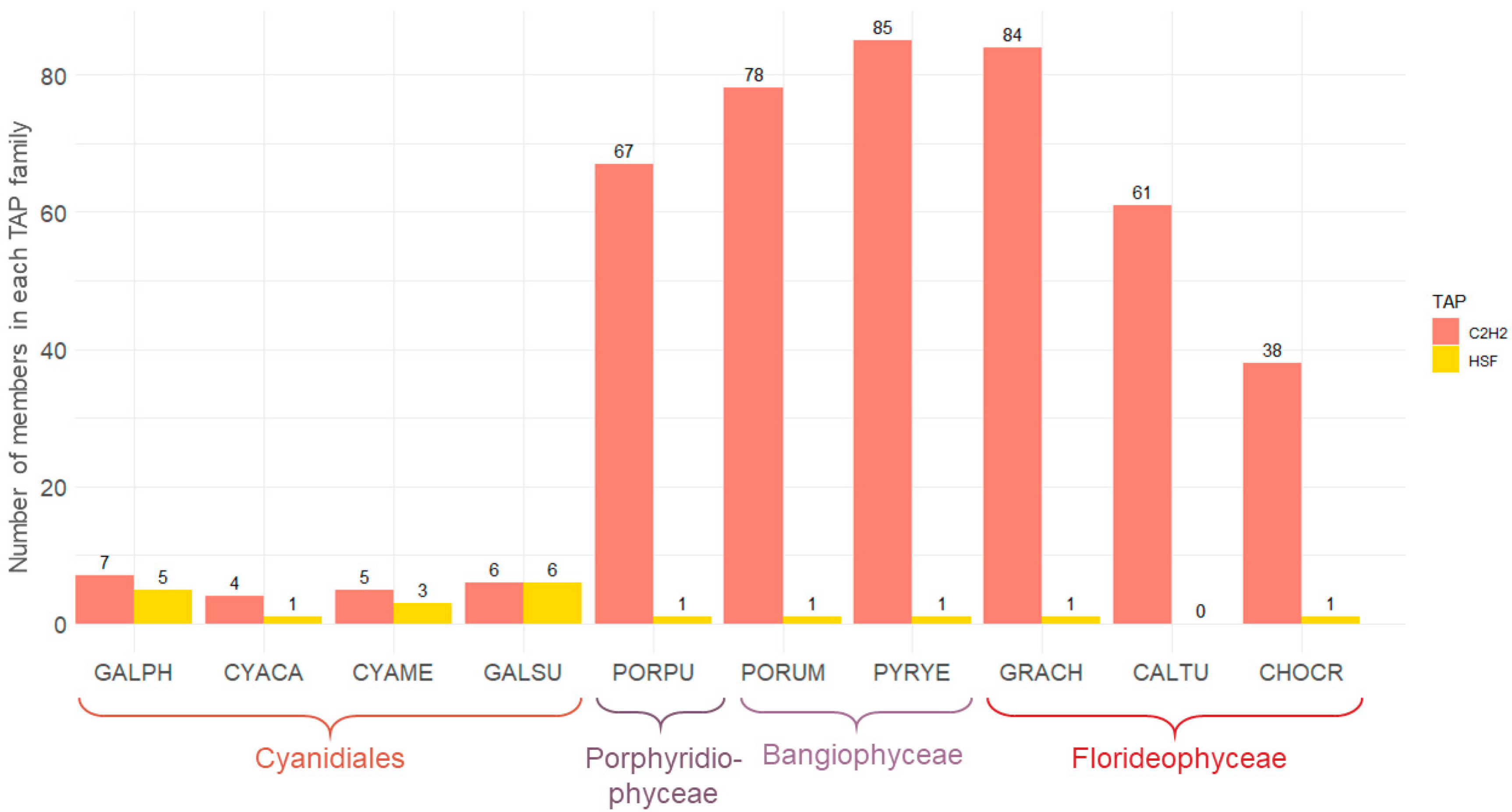
3.2. Differential Occurrence of TAP Families in Unicellular and Multicellular Red Algae
3.3. The Sizes of the C2H2 and HSF Families Differ Significantly across the Red Algae
3.4. Evolution of the TAP Families C2H2 and HSF
3.5. Changes in the Presence and Abundance of TAP Families during Evolution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gould, S.B.; Waller, R.; McFadden, G.I. Plastid Evolution. Annu. Rev. Plant. Biol. 2008, 59, 491–517. [Google Scholar] [CrossRef] [Green Version]
- Strassert, J.F.H.; Irisarri, I.; Williams, T.A.; Burki, F. A molecular timescale for the origin of red algal-derived plastids. Nat. Commun. 2021. [Google Scholar] [CrossRef]
- Fischer, W.W.; Hemp, J.; Johnson, J.E. Evolution of Oxygenic Photosynthesis. Annu. Rev. Earth Planet. Sci. 2016, 44, 647–683. [Google Scholar] [CrossRef]
- Reyes-Prieto, A.; Weber, A.; Bhattacharya, D. The Origin and Establishment of the Plastid in Algae and Plants. Annu. Rev. Genet. 2007, 41, 147–168. [Google Scholar] [CrossRef] [Green Version]
- Lang, D.; Rensing, S.A. The evolution of transcriptional regulation in the viridiplantae and its correlation with morphological complexity. In Evolutionary Transitions to Multicellular Life; Springer: Dordrecht, The Netherlands, 2015. [Google Scholar]
- Cock, J.M.; Collén, J. Independent Emergence of Complex Multicellularity in the Brown and Red Algae. In Evolutionary Transitions to Multicellular Life; Springer: Dordrecht, The Netherlands, 2015. [Google Scholar]
- Collén, J.; Porcel, B.; Carré, W.; Ball, S.; Chaparro, C.; Tonon, T.; Barbeyron, T.; Michel, G.; Noel, B.; Valentin, K.; et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proc. Natl. Acad. Sci. USA 2013, 110, 5247–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, N.J. Bangiomorpha pubescens n. gen., n. sp.: Implications for the evolution of sex, multicellularity, and the Mesoproterozoic/Neoproterozoic radiation of eukaryotes. Paleobiology 2000, 26, 386–404. [Google Scholar] [CrossRef]
- Bengtson, S.; Sallstedt, T.; Belivanova, V.; Whitehouse, M. Three-dimensional preservation of cellular and subcellular structures suggests 1.6 billion-year-old crown-group red algae. PLoS Biol. 2017, 15, e2000735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clerck, O.; Bogaert, K.; Leliaert, F. Diversity and Evolution of Algae. Adv. Bot. Res. 2012, 64, 55–86. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Qiu, H.; Lee, J.; Yoon, H.S.; Weber, A.P.M.; Price, D.C. When Less is More: Red Algae as Models for Studying Gene Loss and Genome Evolution in Eukaryotes. Crit. Rev. Plant. Sci. 2018, 37, 81–99. [Google Scholar] [CrossRef]
- Ciniglia, C.; Yoon, H.S.; Pollio, A.; Pinto, G.; Bhattacharya, D. Hidden biodiversity of the extremophilic Cyanidiales red algae. Mol. Ecol. 2004, 13, 1827–1838. [Google Scholar] [CrossRef]
- Rensing, S.A. Gene duplication as a driver of plant morphogenetic evolution. Curr. Opin. Plant. Biol. 2014, 17, 43–48. [Google Scholar] [CrossRef]
- Lee, J.; Yang, E.C.; Graf, L.; Yang, J.H.; Qiu, H.; Zelzion, U.; Chan, C.X.; Stephens, T.; Weber, A.P.M.; Boo, G.H.; et al. Analysis of the Draft Genome of the Red Seaweed Gracilariopsis chorda Provides Insights into Genome Size Evolution in Rhodophyta. Mol. Biol. Evol. 2018, 35, 1869–1886. [Google Scholar] [CrossRef] [Green Version]
- Seckbach, J.; Chapman, D.J. (Eds.) Red Algae in the Genomic Age; Cellular Origin, Life in Extreme Habitats and Astrobiology 13; Springer: Dordrecht, The Netherlands, 2010; pp. 25–42. [Google Scholar] [CrossRef]
- Lang, D.; Weiche, B.; Timmerhaus, G.; Richardt, S.; Pachón, D.M.R.; Corrêa, L.G.G.; Reski, R.; Mueller-Roeber, B.; Rensing, S.A. Genome-Wide Phylogenetic Comparative Analysis of Plant Transcriptional Regulation: A Timeline of Loss, Gain, Expansion, and Correlation with Complexity. Genome Biol. Evol. 2010, 2, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsson, P.K.; Mühlich, C.; Ullrich, K.; Rensing, S.A. Comprehensive Genome-Wide Classification Reveals That Many Plant-Specific Transcription Factors Evolved in Streptophyte Algae. Genome Biol. Evol. 2017, 9, 3384–3397. [Google Scholar] [CrossRef] [PubMed]
- Richardt, S.; Lang, D.; Reski, R.; Frank, W.; Rensing, S.A. PlanTAPDB, a Phylogeny-Based Resource of Plant Transcription-Associated Proteins. Plant. Physiol. 2007, 143, 1452–1466. [Google Scholar] [CrossRef] [Green Version]
- Albertin, C.; Simakov, O.; Mitros, T.; Wang, Z.Y.; Pungor, J.R.; Edsinger-Gonzales, E.; Brenner, S.; Ragsdale, C.W.; Rokhsar, D.S. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nat. Cell Biol. 2015, 524, 220–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, C.; Sørensen, I.; Sun, X.; Sun, H.; Behar, H.; Alseekh, S.; Philippe, G.; Lopez, K.P.; Sun, L.; Reed, R.; et al. The Penium margaritaceum Genome: Hallmarks of the Origins of Land Plants. Cell 2020, 181, 1097–1111.e12. [Google Scholar] [CrossRef] [PubMed]
- De Clerck, O.; Kao, S.-M.; Bogaert, K.; Blomme, J.; Foflonker, F.; Kwantes, M.; Vancaester, E.; Vanderstraeten, L.; Aydogdu, E.; Boesger, J.; et al. Insights into the Evolution of Multicellularity from the Sea Lettuce Genome. Curr. Biol. 2018, 28, 2921–2933.e5. [Google Scholar] [CrossRef] [Green Version]
- Bowman, J.L.; Kohchi, T.; Yamato, K.T.; Jenkins, J.; Shu, S.; Ishizaki, K.; Yamaoka, S.; Nishihama, R.; Nakamura, Y.; Berger, F.; et al. Insights into Land Plant Evolution Garnered from the Marchantia polymorpha Genome. Cell 2017, 171, 287–304.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puttick, M.; Morris, J.L.; Williams, T.; Cox, C.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Schneider, H.; Pisani, D.; et al. The Interrelationships of Land Plants and the Nature of the Ancestral Embryophyte. Curr. Biol. 2018, 28, 733–745.e2. [Google Scholar] [CrossRef] [Green Version]
- Adl, S.M.; Bass, D.; Lane, C.; Lukeš, J.; Schoch, C.L.; Smirnov, A.; Agatha, S.; Berney, C.; Brown, M.; Burki, F.; et al. Revisions to the Classification, Nomenclature, and Diversity of Eukaryotes. J. Eukaryot. Microbiol. 2019, 66, 4–119. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Gómez, S.A.; Mejía-Franco, F.G.; Durnin, K.; Colp, M.; Grisdale, C.J.; Archibald, J.M.; Slamovits, C.H. The New Red Algal Subphylum Proteorhodophytina Comprises the Largest and Most Divergent Plastid Genomes Known. Curr. Biol. 2017, 27, 1677–1684.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Initiative, O.T.P.T. One thousand plant transcriptomes and the phylogenomics of green plants. Nat. Cell Biol. 2019, 574, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Sterck, L.; Billiau, K.; Abeel, T.; Rouzé, P.; Van de Peer, Y. ORCAE: Online resource for community annotation of eukaryotes. Nat. Methods 2012, 9, 1041. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Seqtk, Toolkit for Processing Sequences in FASTA/Q Formats. Available online: https://github.com/lh3/seqtk (accessed on 16 March 2021).
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Eddy, S.R. HMMER, version 3.3. Available online: hmmer.org (accessed on 10 November 2020).
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Englbrecht, C.C.; Schoof, H.; Böhm, S. Conservation, diversification and expansion of C2H2 zinc finger proteins in the Arabidopsis thaliana genome. BMC Genom. 2004, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Dröge-Laser, W.; Snoek, B.L.; Snel, B.; Weiste, C. The Arabidopsis bZIP transcription factor family—An update. Curr. Opin. Plant. Biol. 2018, 45, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lv, W.; Zhang, H.; Ma, L.; Li, P.; Ge, L.; Li, G. Genome-wide analysis of the basic Helix-Loop-Helix (bHLH) transcription factor family in maize. BMC Plant Biol. 2018, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org (accessed on 8 February 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Vu, V.Q. ggbiplot: A ggplot2 Based Biplot, Version 0.55. Available online: http://github.com/vqv/ggbiplot (accessed on 8 February 2021).
- Csurös, M. Count: Evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 2010, 26, 1910–1912. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Price, D.C.; Weber, A.P.; Reeb, V.; Yang, E.C.; Lee, J.M.; Kim, S.Y.; Yoon, H.S.; Bhattacharya, D. Adaptation through horizontal gene transfer in the cryptoendolithic red alga Galdieria phlegrea. Curr. Biol. 2013, 23, R865–R866. [Google Scholar] [CrossRef] [Green Version]
- Miyagishima, S.; Wei, J.L.; Nozaki, H.; Hirooka, S. Cyanidiales: Evolution and habitats. In Cyanidioschyzon merolae: A New Model Eukaryote for Cell and Organelle Biology; Springer: Berlin, Germany, 2018; pp. 3–15. ISBN 9789811061011. [Google Scholar]
- Waterhouse, A.M.; Procter, J.; Martin, D.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Frickenhaus, S.; Beszteri, B. Stephan Frickenhaus & Bánk Beszteri August 11, 2009. Mol. Biol. Evol. 2009, 6–7. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Hoehna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- FigTree v1.4.4. Available online: https://github.com/rambaut/figtree (accessed on 31 March 2021).
- Chen, J.; Yu, J.; Ge, L.; Wang, H.; Berbel, A.; Liu, Y.; Chen, Y.; Li, G.; Tadege, M.; Wen, J.; et al. Control of dissected leaf morphology by a Cys(2)His(2) zinc finger transcription factor in the model legume Medicago truncatula. Proc. Natl. Acad. Sci. USA 2010, 107, 10754–10759. [Google Scholar] [CrossRef] [Green Version]
- Ciftci-Yilmaz, S.; Mittler, R. The zinc finger network of plants. Cell. Mol. Life Sci. 2008, 65, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Tadepally, H.D.; Burger, G.; Aubry, M. Evolution of C2H2-zinc finger genes and subfamilies in mammals: Species-specific duplication and loss of clusters, genes and effector domains. BMC Evol. Biol. 2008, 8, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seetharam, A.; Stuart, G.W. A study on the distribution of 37 well conserved families of C2H2 zinc finger genes in eukaryotes. BMC Genom. 2013, 14, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C. Heat Shock Transcription Factors: Structure and Regulation. Annu. Rev. Cell Dev. Biol. 1995, 11, 441–469. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Sakayama, H.; de Vries, J.; Buschmann, H.; Saint-Marcoux, D.; Ullrich, K.K.; Haas, F.; Vanderstraeten, L.; Becker, D.; Lang, D.; et al. The Chara Genome: Secondary Complexity and Implications for Plant Terrestrialization. Cell 2018, 174, 448–464.e24. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Marín, B.; Rakijas, J.B.; Tyagi, A.; Pandey, A.; Hanschen, E.R.; Anderson, J.; Heffel, M.G.; Platt, T.G.; Olson, B.J.S.C. Gene loss during the transition to multicellularity. bioRxiv Prepr. 2021, 6. [Google Scholar] [CrossRef]
- de Vries, J.; Gould, S.B. The monoplastidic bottleneck in algae and plant evolution. J. Cell Sci. 2018, 131, jcs.203414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, C.; Nelson-Sathi, S.; Roettger, M.; Sousa, F.L.; Lockhart, P.J.; Bryant, D.; Hazkani-Covo, E.; McInerney, J.O.; Landan, G.; Martin, W.F. Endosymbiotic origin and differential loss of eukaryotic genes. Nat. Cell Biol. 2015, 524, 427–432. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petroll, R.; Schreiber, M.; Finke, H.; Cock, J.M.; Gould, S.B.; Rensing, S.A. Signatures of Transcription Factor Evolution and the Secondary Gain of Red Algae Complexity. Genes 2021, 12, 1055. https://doi.org/10.3390/genes12071055
Petroll R, Schreiber M, Finke H, Cock JM, Gould SB, Rensing SA. Signatures of Transcription Factor Evolution and the Secondary Gain of Red Algae Complexity. Genes. 2021; 12(7):1055. https://doi.org/10.3390/genes12071055
Chicago/Turabian StylePetroll, Romy, Mona Schreiber, Hermann Finke, J. Mark Cock, Sven B. Gould, and Stefan A. Rensing. 2021. "Signatures of Transcription Factor Evolution and the Secondary Gain of Red Algae Complexity" Genes 12, no. 7: 1055. https://doi.org/10.3390/genes12071055
APA StylePetroll, R., Schreiber, M., Finke, H., Cock, J. M., Gould, S. B., & Rensing, S. A. (2021). Signatures of Transcription Factor Evolution and the Secondary Gain of Red Algae Complexity. Genes, 12(7), 1055. https://doi.org/10.3390/genes12071055