
Genetic and Transcriptomic Analysis Reveal the Molecular Basis of Photoperiod-Regulated Flowering in Xishuangbanna Cucumber (Cucumis sativus L. var. xishuangbannesis Qi et Yuan)



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and SSR-Based Mapping Analysis
2.2. QTL-Seq Analysis
2.3. Prediction and Verification of Candidate Genes Regulating DFF
2.4. Plant Materials and Growth Conditions of RNA-Seq Analysis
2.5. RNA Extraction, Library Preparation and Sequencing
2.6. RNA-Seq Data and Enrichment Analysis
2.7. Quantitative Real-Time PCR Analysis of RNA-Seq Data
2.8. Assay of Phenotypic and Physiological Parameters
2.9. Weighted Gene Co-Expression Network Analysis
3. Results
3.1. Both Vegetative and Reproductive Growth Were Regulated by Photoperiod in XIS Cucumber
3.2. Stable QTL Loci Were Obtained on DFF by the SSR-Based Mapping
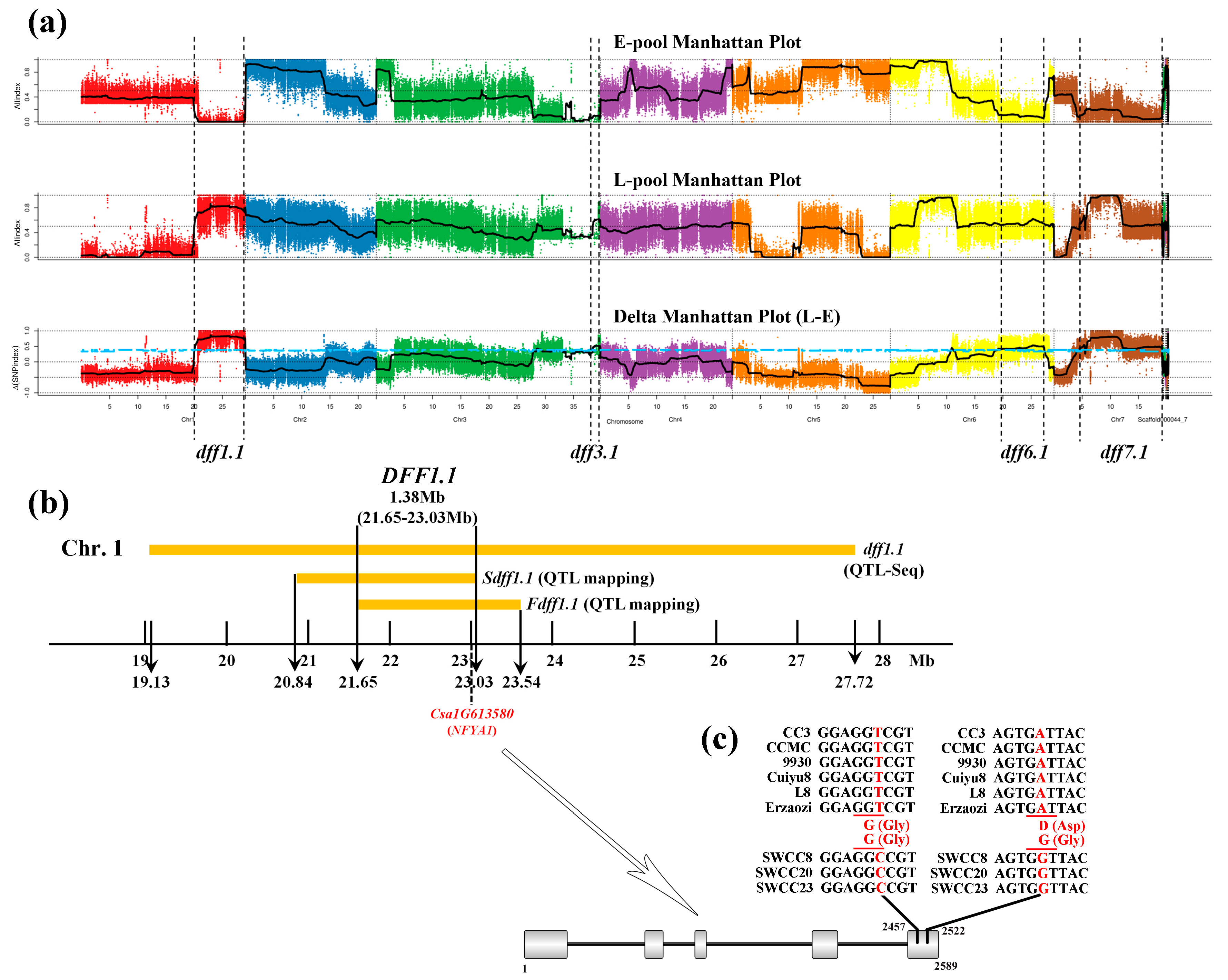
3.3. DFF1.1 Is a Major Effect QTL Controlling Photoperiod Responsive Flowering
3.4. The Response of Hormone Was Significantly Different under SD and ED Regimes
3.5. The SD Condition Accelerated the Accumulation of Carbohydrates
3.6. Flowering Related Genes and Transcription Factors Showed Different Expression Patterns in Response to SD and ED Photoperiod Regimes
3.7. A Co-Expression Network Was Constructed by CsaNFYA1 and Related Genes
4. Discussion
4.1. QTLs on Flowering Time Are Conservative in Photoperiod-Sensitive Cucumbers
4.2. The Effect of Photoperiod on Flowering Process Is Different in Time
4.3. Various Types of Genes Are Involved in the Regulation of Photoperiodic Flowering
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amasino, R.M.; Michaels, S.D. The timing of flowering. Plant Physiol. 2010, 154, 516–520. [Google Scholar] [CrossRef]
- Song, Y.H.; Shim, J.S.; Kinmonth-Schultz, H.A.; Imaizumi, T. Photoperiodic flowering: Time measurement mechanisms in leaves. Annu. Rev. Plant Biol. 2015, 66, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Koornneef, M.; Alonso-Blanco, C.; Peeters, A.J.M.; Soppe, W. Genetic control of flowering time in Arabidopsis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 345–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayama, R.; Yokoi, S.; Tamaki, S.; Yano, M.; Shimamoto, K. Adaptation of photoperiodic control pathways produces short-day flowering in rice. Nature 2003, 422, 719–722. [Google Scholar] [CrossRef]
- Weller, J.L.; Vander Schoor, J.K.; Perez-Wright, E.C.; Hecht, V.; Gonzalez, A.M.; Capel, C.; Yuste-Lisbona, F.J.; Lozano, R.; Santalla, M. Parallel origins of photoperiod adaptation following dual domestications of common bean. J. Exp. Bot. 2019, 70, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Corbesier, L.; Vincent, C.; Jang, S.H.; Fornara, F.; Fan, Q.Z.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, S.; Matsuo, S.; Wong, H.L.; Yokoi, S.; Shimamoto, K. Hd3a protein is a mobile flowering signal in rice. Science 2007, 316, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Putterill, J.; Robson, F.; Lee, K.; Simon, R.; Coupland, G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 1995, 80, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, D.; Kong, F.Y.; Lin, K.; Zhang, H.S.; Li, G. The Arabidopsis thaliana Nuclear Factor Y transcription factors. Front. Plant Sci. 2017, 7, 2045. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef]
- Gao, H.; Jin, M.N.; Zheng, X.M.; Chen, J.; Yuan, D.Y.; Xin, Y.Y.; Wang, M.Q.; Huang, D.Y.; Zhang, Z.; Zhou, K.N.; et al. Days to heading 7, a major quantitative locus determining photoperiod sensitivity and regional adaptation in rice. Proc. Natl. Acad. Sci. USA 2014, 111, 16337–16342. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Buylla, E.R.; Benitez, M.; Corvera-Poire, A.; Chaos Cador, A.; de Folter, S.; Gamboa de Buen, A.; Garay-Arroyo, A.; Garcia-Ponce, B.; Jaimes-Miranda, F.; Perez-Ruiz, R.V.; et al. Flower development. Arab. Book 2010, 8, e0127. [Google Scholar] [CrossRef] [Green Version]
- Qi, C.Z.; Yuan, Z.Z.; Li, Y.X. A new type of cucumber-Xishuangbanna cucumber. Acta Hortic. Sin. 1983, 10, 259–264. [Google Scholar]
- Chen, J.F.; Zhang, S.L.; Zhang, X.G. The Xishuangbanna gourd (Cucumis sativus L. var. xishuangbannanesis Qi et Yuan), a traditionally cultivated plant of the Hanai people, Xishuangbanna, Yunnan, China. Cucurbit Genet. Coop. Rep. 1994, 17, 18–20. [Google Scholar]
- Shen, D.; Li, X.X.; Song, J.P.; Wang, H.P.; Qiu, Y. Effects of different sowing dates on sex type and lateral stem development of Xishuangbanna cucumber. China Veget. 2011, 6, 22–27. [Google Scholar]
- Bo, K.L.; Chen, L.Z.; Qian, C.T.; Zhang, S.X.; Chen, J.F. Short-day treatments induce flowering of Xishuangbanna cucumber. China Cucurbits Veg. 2010, 23, 1–3. [Google Scholar]
- Bo, K.L.; Ma, Z.; Chen, J.F.; Weng, Y.Q. Molecular mapping reveals structural rearrangements and quantitative trait loci underlying traits with local adaptation in semi-wild Xishuangbanna cucumber (Cucumis sativus L. var. xishuangbannanesis Qi et Yuan). Theor. Appl. Genet. 2015, 128, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.B. Precision mapping of quantitative trait loci. Genetics 1994, 136, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.D.C.E.; Wang, S.C.; Zeng, Z.B. Composite interval mapping and multiple interval mapping: Procedures and guidelines for using Windows QTL Cartographer. Methods Mol. Biol. 2012, 871, 75–119. [Google Scholar]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.I.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malatesta, M. Histological and histochemical methods—Theory and practice. Eur. J. Histochem. 2016, 60. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rosa, M.; Hilal, M.; Gonzalez, J.A.; Prado, F.E. Low-temperature effect on enzyme activities involved in sucrose-starch partitioning in salt-stressed and salt-acclimated cotyledons of quinoa (Chenopodium quinoa Willd.) seedlings. Plant Physiol. Biochem. 2009, 47, 300–307. [Google Scholar] [CrossRef]
- Yano, M.; Katayose, Y.; Ashikari, M.; Yamanouchi, U.; Monna, L.; Fuse, T.; Baba, T.; Yamamoto, K.; Umehara, Y.; Nagamura, Y.; et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 2000, 12, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.P.; Qu, S.P.; Bo, K.L.; Gao, M.L.; Haider, K.R.; Weng, Y.Q. QTL mapping of domestication and diversifying selection related traits in round-fruited semi-wild Xishuangbanna cucumber (Cucumis sativus L. var. xishuangbannanesis). Theor. Appl. Genet. 2017, 130, 1531–1548. [Google Scholar] [CrossRef]
- Sheng, Y.Y.; Pan, Y.P.; Li, Y.H.; Yang, L.M.; Weng, Y.Q. Quantitative trait loci for fruit size and flowering time-related traits under domestication and diversifying selection in cucumber (Cucumis sativus). Plant Breed. 2020, 139, 176–191. [Google Scholar] [CrossRef]
- Nardini, M.; Gnesutta, N.; Donati, G.; Gatta, R.; Forni, C.; Fossati, A.; Vonrhein, C.; Moras, D.; Romier, C.; Bolognesi, M.; et al. Sequence-specific transcription factor NF-Y displays histone-like DNA binding and H2B-like ubiquitination. Cell 2013, 152, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Siriwardana, C.L.; Gnesutta, N.; Kumimoto, R.W.; Jones, D.S.; Myers, Z.A.; Mantovani, R.; Holt, B.F. NUCLEAR FACTOR Y, subunit A (NF-YA) proteins positively regulate flowering and act through FLOWERING LOCUS T. PLoS Genet. 2016, 12, e1006496. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef]
- Ren, L.P.; Liu, T.; Cheng, Y.; Sun, J.; Gao, J.J.; Dong, B.; Chen, S.M.; Chen, F.D.; Jiang, J.F. Transcriptomic analysis of differentially expressed genes in the floral transition of the summer flowering chrysanthemum. BMC Genom. 2016, 17, 673. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.P.; Li, Y.; Pan, J.J.; Lou, D.J.; Hu, Y.R.; Yu, D.Q. The bHLH transcription factors MYC2, MYC3, and MYC4 are required for jasmonate-mediated inhibition of flowering in Arabidopsis. Mol. Plant 2017, 10, 1461–1464. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Guan, Y.X.; Liu, Y.N.; Zhang, Z.H.; Jaffar, M.A.; Song, A.P.; Chen, S.M.; Jiang, J.F.; Chen, F.D. Regulation of flowering time in chrysanthemum by the R2R3 MYB transcription factor CmMYB2 is associated with changes in gibberellin metabolism. Hortic. Res. 2020, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Krizek, B.A. AINTEGUMENTA and AINTEGUMENTA-LIKE6 act redundantly to regulate Arabidopsis floral growth and patterning. Plant Physiol. 2009, 150, 1916–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, F.W.; Zhang, C.Q.; Jiang, X.Q.; Kang, M.; Yin, X.; Lu, P.T.; Zhang, X.; Zheng, Y.; Gao, J.P. RhNAC2 and RhEXPA4 are involved in the regulation of dehydration tolerance during the expansion of rose petals. Plant Physiol. 2012, 160, 2064–2082. [Google Scholar] [CrossRef] [Green Version]
- Krizek, B.A. Auxin regulation of Arabidopsis flower development involves members of the AINTEGUMENTA-LIKE/PLETHORA (AIL/PLT) family. J. Exp. Bot. 2011, 62, 3311–3319. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Wu, M.F.; Winter, C.M.; Berns, M.C.; Nole-Wilson, S.; Yamaguchi, A.; Coupland, G.; Krizek, B.A.; Wagner, D. A molecular framework for auxin-mediated initiation of flower primordia. Dev. Cell 2013, 24, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, N.; Khan, N.A.; Ferrante, A.; Trivellini, A.; Francini, A.; Khan, M.I.R. Ethylene role in plant growth, development and senescence: Interaction with other phytohormones. Front. Plant Sci. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.N.; Cate, J.H.D. The structure and function of the eukaryotic ribosome. Cold Spring Harb. Perspect. Biol. 2012, 4, a011536. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, N.; Kita, M.; Niinuma, K.; Ito, S.; Yamashino, T.; Mizoguchi, T.; Mizuno, T. Arabidopsis clock-associated pseudo-response regulators PRR9, PRR7 and PRR5 coordinately and positively regulate flowering time through the canonical CONSTANS-dependent photoperiodic pathway. Plant Cell Physiol. 2007, 48, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.M.; Austin, R.S.; Blanvillain-Baufume, S.; Reback, M.A.; Monniaux, M.; Wu, M.F.; Sang, Y.; Yamaguchi, A.; Yamaguchi, N.; Parker, J.E.; et al. LEAFY target genes reveal floral regulatory logic, cis motifs, and a link to biotic stimulus response. Dev. Cell 2011, 20, 430–443. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.H.; Muino, J.M.; Blanvillain, R.; Busscher, M.; Busscher-Lange, J.; Dinh, Q.D.; Liu, S.J.; Westphal, A.H.; Boeren, S.; et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad. Sci. USA 2012, 109, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.B.; Chen, Y.H.; Wang, Z.Y.; Chen, Z.L.; Gu, H.Y.; Qu, L.J. Constitutive expression of CIR1 (RVE2) affects several circadian-regulated processes and seed germination in Arabidopsis. Plant J. 2007, 51, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; Cheng, F.F.; Sun, Z.H.; Tang, J.H.; Wu, L.C.; Ku, L.X.; Chen, Y.H. Genetic analysis of photoperiod sensitivity in a tropical by temperate maize recombinant inbred population using molecular markers. Theor. Appl. Genet. 2008, 117, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Kamran, A.; Iqbal, M.; Navabi, A.; Randhawa, H.; Pozniak, C.; Spaner, D. Earliness per se QTLs and their interaction with the photoperiod insensitive allele Ppd-D1a in the Cutler x AC Barrie spring wheat population. Theor. Appl. Genet. 2013, 126, 1965–1976. [Google Scholar] [CrossRef]
- Matsubara, K.; Kono, I.; Hori, K.; Nonoue, Y.; Ono, N.; Shomura, A.; Mizubayashi, T.; Yamamoto, S.; Yamanouchi, U.; Shirasawa, K.; et al. Novel QTLs for photoperiodic flowering revealed by using reciprocal backcross inbred lines from crosses between japonica rice cultivars. Theor. Appl. Genet. 2008, 117, 935–945. [Google Scholar] [CrossRef]
- Thomas, B.; Vince-Prue, D. Photoperiodic Control of Flower Initiation. In Photoperiodism in Plants, 2nd ed.; Academic Press: London, UK, 1997; pp. 3–257. [Google Scholar]
- Jeong, S.; Clark, S.E. Photoperiod regulates flower meristem development in Arabidopsis thaliana. Genetics 2005, 169, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Yuan, W.Y.; Tian, S.; Zheng, Q.J.; He, Y.H. SIN3 LIKE genes mediate long-day induction of flowering but inhibit the floral transition in short days through histone deacetylation in Arabidopsis. Plant J. 2019, 100, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Acock, M.C.; Acock, B. Photoperiod sensitivity during flower development of opium poppy (Papaver somniferum L.). Ann. Bot. 1997, 79, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Li, H.B.; Li, Y.Y.; Li, Z.; Qi, J.J.; Lin, T.; Yang, X.Y.; Zhang, Z.H.; Huang, S.W. FLOWERING LOCUS T improves cucumber adaptation to higher latitudes. Plant Physiol. 2020, 182, 908–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Lithy, M.E.; Reymond, M.; Stich, B.; Koornneef, M.; Vreugdenhil, D. Relation among plant growth, carbohydrates and flowering time in the Arabidopsis Landsberg erecta x Kondara recombinant inbred line population. Plant Cell Environ. 2010, 33, 1369–1382. [Google Scholar] [CrossRef]
- Fornara, F.; Coupland, G. Plant phase transitions make a SPLash. Cell 2009, 138, 625–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaller, G.E. Ethylene and the regulation of plant development. BMC Biol. 2012, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, R. The molecular biology of the CCAAT-binding factor NF-Y. Gene 1999, 239, 15–27. [Google Scholar] [CrossRef]
- Sinha, S.; Maity, S.N.; Lu, J.; de Crombrugghe, B. Recombinant rat CBF-C, the third subunit of CBF/NFY, allows formation of a protein-DNA complex with CBF-A and CBF-B and with yeast HAP2 and HAP3. Proc. Natl. Acad. Sci. USA 1995, 92, 1624–1628. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Kim, I.S.; Sohn, K.Y.; de Crombrugghe, B.; Maity, S.N. Three classes of mutations in the A subunit of the CCAAT-binding factor CBF delineate functional domains involved in the three-step assembly of the CBF-DNA complex. Mol. Cell Biol. 1996, 16, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Ben-Naim, O.; Eshed, R.; Parnis, A.; Teper-Bamnolker, P.; Shalit, A.; Coupland, G.; Samach, A.; Lifschitz, E. The CCAAT binding factor can mediate interactions between CONSTANS-like proteins and DNA. Plant J. 2006, 46, 462–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumimoto, R.W.; Adam, L.; Hymus, G.J.; Repetti, P.P.; Reuber, T.L.; Marion, C.M.; Hempel, F.D.; Ratcliffe, O.J. The Nuclear Factor Y subunits NF-YB2 and NF-YB3 play additive roles in the promotion of flowering by inductive long-day photoperiods in Arabidopsis. Planta 2008, 228, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Kumimoto, R.W.; Gnesutta, N.; Calogero, A.M.; Mantovani, R.; Holt, B.F. A distal CCAAT/NUCLEAR FACTOR Y complex promotes chromatin looping at the FLOWERING LOCUS T promoter and regulates the timing of flowering in Arabidopsis. Plant Cell 2014, 26, 1009–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenkel, S.; Turck, F.; Singer, K.; Gissot, L.; Le Gourrierec, J.; Samach, A.; Coupland, G. CONSTANS and the CCAAT box binding complex share a functionally important domain and interact to regulate flowering of Arabidopsis. Plant Cell 2006, 18, 2971–2984. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.B.; Zhang, D.; Li, Y.M.; Shen, Y.W.; Zhao, C.P.; Ma, J.J.; An, N.; Han, M.Y. Transcription profiles reveal sugar and hormone signaling pathways mediating flower induction in Apple (Malus domestica Borkh.). Plant Cell Physiol. 2015, 56, 2052–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
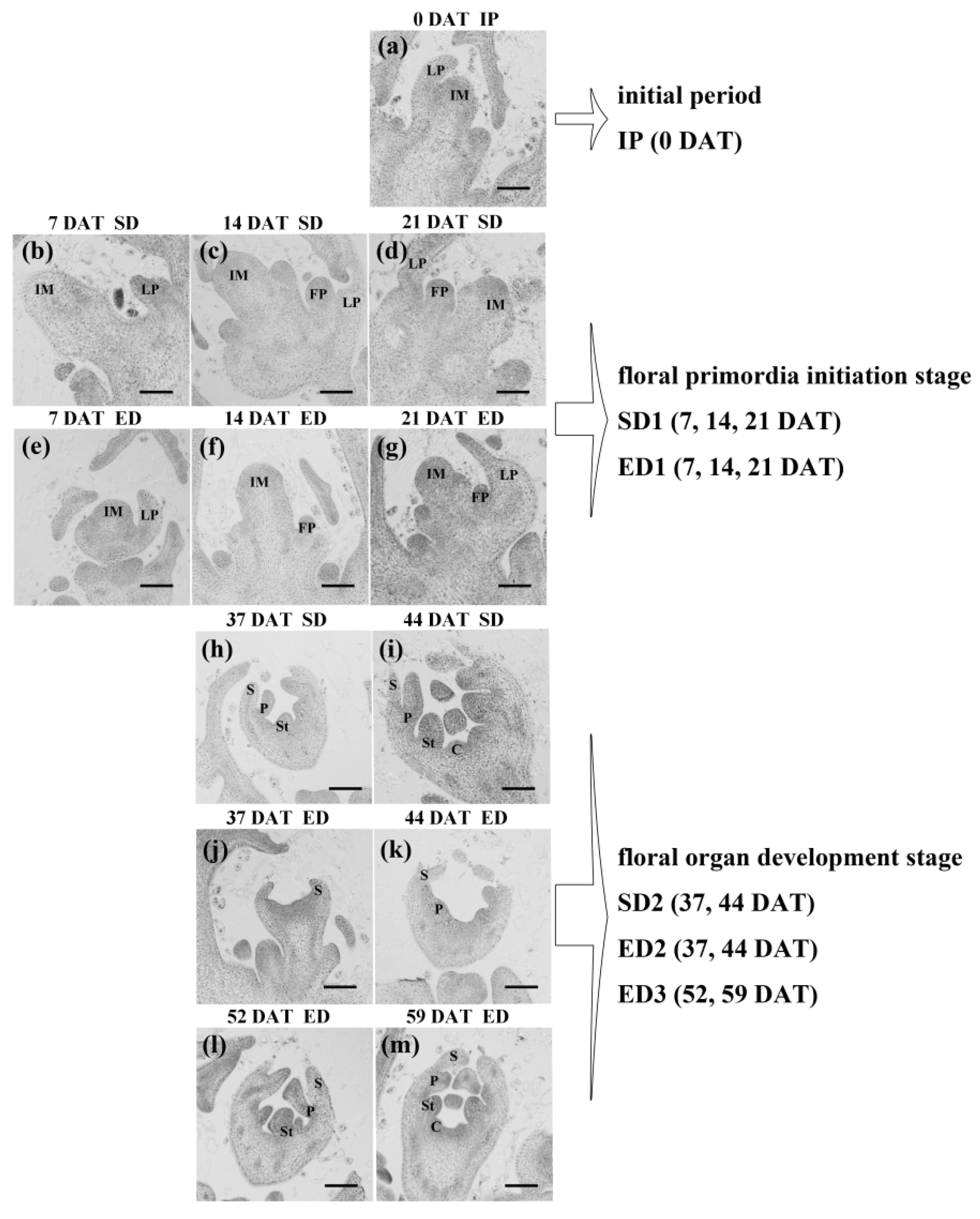
| Photoperiod Regimes | Initial Period | Floral Primordial Initiation Stage | Floral Organ Development Stage | |
|---|---|---|---|---|
| SD (8 h/16 h) | IP (0 DAT) | SD1 (7, 14, 21 DAT) | SD2 (37, 44 DAT) | |
| ED (12 h/12 h) | ED1 (7, 14, 21 DAT) | ED2 (37, 44 DAT) | ED3 (52, 59 DAT) | |
| Treatments (Day/Night) | DFF (d) | NFF | ph (cm) |
|---|---|---|---|
| SD (8 h/16 h) | 80.0 ± 0.0 b | 11.3 ± 2.3 b | 55.5 ± 2.5 b |
| ED (12 h/12 h) | 98.0 ± 0.0 a | 16.0 ± 0.0 a | 68.2 ± 2.4 a |
| Seasons | Parents | RIL Populations | ||||
|---|---|---|---|---|---|---|
| CC3 | SWCC8 | Range | Mean ± SD | Kurtosis | Skewness | |
| 2016 spring | 47.2 ± 0.9 | 87.8 ± 0.6 | 45.0~90.3 | 62.8 ± 0.7 | 0.28 | 0.32 |
| 2016 fall | 32.5 ± 0.6 | 69.5 ± 0.9 | 32.0~55.0 | 42.5 ± 0.5 | −1.19 | 0.01 |
| QTL Loci | Chr. | LOD Score | R2 (%) | Additive Effects | Peak Location (cM) | Marker Interval |
|---|---|---|---|---|---|---|
| Sdff1.1 | 1 | 3.5 | 10.6 | −7.9 | 86.2 | SSR16841-SSR23049 |
| Fdff1.1 | 1 | 4.9 | 21.7 | −3.6 | 88.8 | SSR05723-SSR22638 |
| Number | QTL Loci | Chr. | Parental Lines (Left, Female Parent; Right, Male Parent) | References | |
|---|---|---|---|---|---|
| 1 | dff1.1 | 1 | CC3 (cultivated cucumber, insensitive) | SWCC8 (semi-wild XIS cucumber, sensitive) | QTL-Seq results in this study (from 1 to 4) |
| 2 | dff3.1 | 3 | |||
| 3 | dff6.1 | 6 | |||
| 4 | dff7.1 | 7 | |||
| 5 | Sdff1.1 | 1 | SSR-based mapping results in this study (from 5 to 6) | ||
| 6 | Fdff1.1 | 1 | |||
| 7 | ft1.1 | 1 | Gy14 (cultivated cucumber, insensitive) | WI7221 (wild cucumber, sensitive) | Sheng et al. [31] (from 7 to 10) |
| 8 | ft1.1 | 1 | |||
| 9 | ft6.3 | 6 | |||
| 10 | ft6.3 | 6 | |||
| 11 | fft1.1 | 1 | WI7176 (semi-wild XIS cucumber, sensitive) | WI7200 (landrace originally collected from Thailand, insensitive) | Pan et al. [30] (from 11 to 15) |
| 12 | mft1.1 | 1 | |||
| 13 | fft5.1 | 5 | |||
| 14 | fft6.2 | 6 | |||
| 15 | mft6.2 | 6 | |||
| 16 | fft1.1 | 1 | CC3 (cultivated cucumber, insensitive) | SWCC8 (semi-wild XIS cucumber, sensitive) | Bo et al. [17] (from 16 to 17) |
| 17 | fft6.1 | 6 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Z.; Jahn, M.; Qin, X.; Obel, H.O.; Yang, F.; Li, J.; Chen, J. Genetic and Transcriptomic Analysis Reveal the Molecular Basis of Photoperiod-Regulated Flowering in Xishuangbanna Cucumber (Cucumis sativus L. var. xishuangbannesis Qi et Yuan). Genes 2021, 12, 1064. https://doi.org/10.3390/genes12071064
Tian Z, Jahn M, Qin X, Obel HO, Yang F, Li J, Chen J. Genetic and Transcriptomic Analysis Reveal the Molecular Basis of Photoperiod-Regulated Flowering in Xishuangbanna Cucumber (Cucumis sativus L. var. xishuangbannesis Qi et Yuan). Genes. 2021; 12(7):1064. https://doi.org/10.3390/genes12071064
Chicago/Turabian StyleTian, Zhen, Molly Jahn, Xiaodong Qin, Hesbon Ochieng Obel, Fan Yang, Ji Li, and Jinfeng Chen. 2021. "Genetic and Transcriptomic Analysis Reveal the Molecular Basis of Photoperiod-Regulated Flowering in Xishuangbanna Cucumber (Cucumis sativus L. var. xishuangbannesis Qi et Yuan)" Genes 12, no. 7: 1064. https://doi.org/10.3390/genes12071064
APA StyleTian, Z., Jahn, M., Qin, X., Obel, H. O., Yang, F., Li, J., & Chen, J. (2021). Genetic and Transcriptomic Analysis Reveal the Molecular Basis of Photoperiod-Regulated Flowering in Xishuangbanna Cucumber (Cucumis sativus L. var. xishuangbannesis Qi et Yuan). Genes, 12(7), 1064. https://doi.org/10.3390/genes12071064