
Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder


Abstract
:1. Introduction
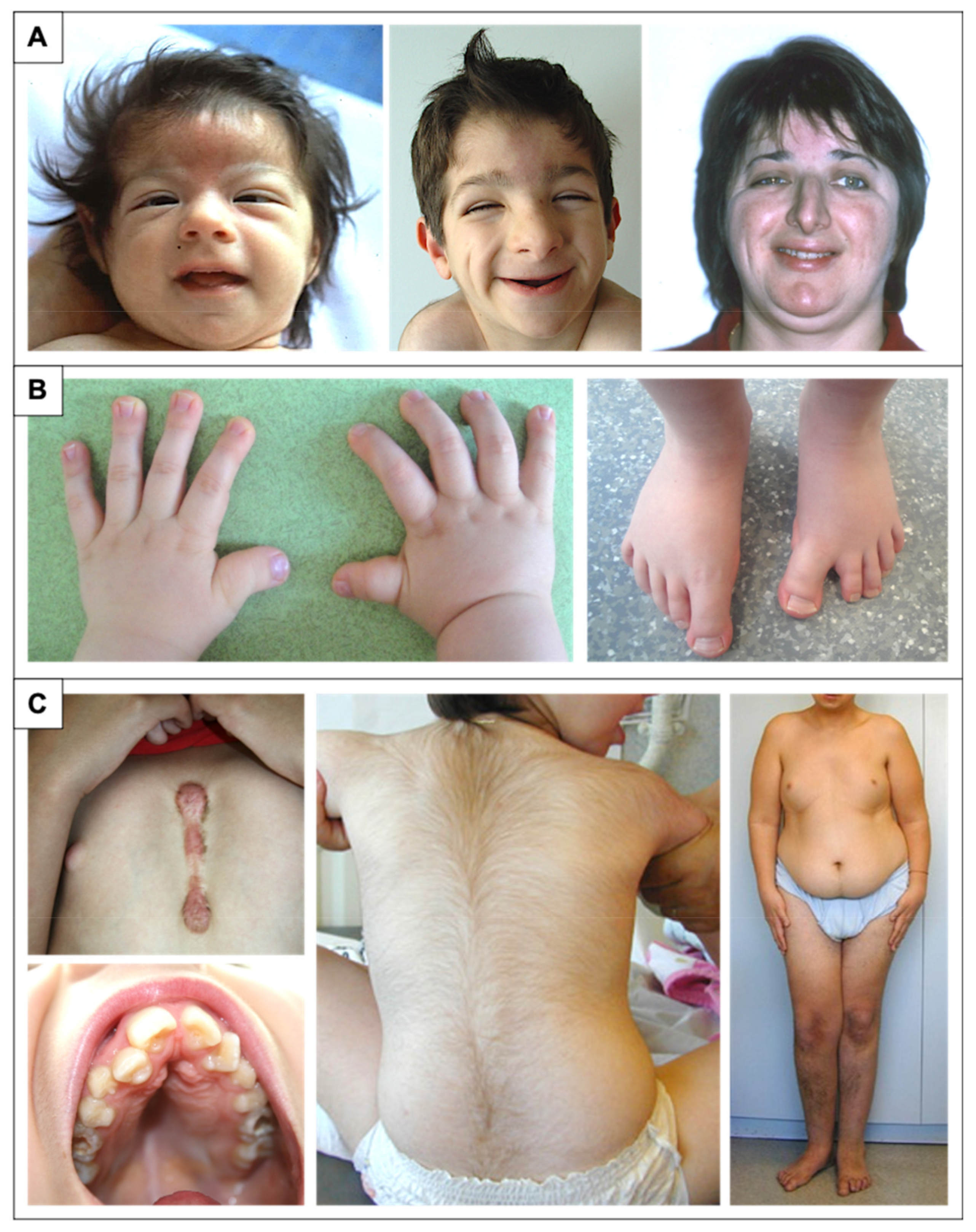
2. Clinical Description
2.1. Antenatal Anomalies and Pregnancy
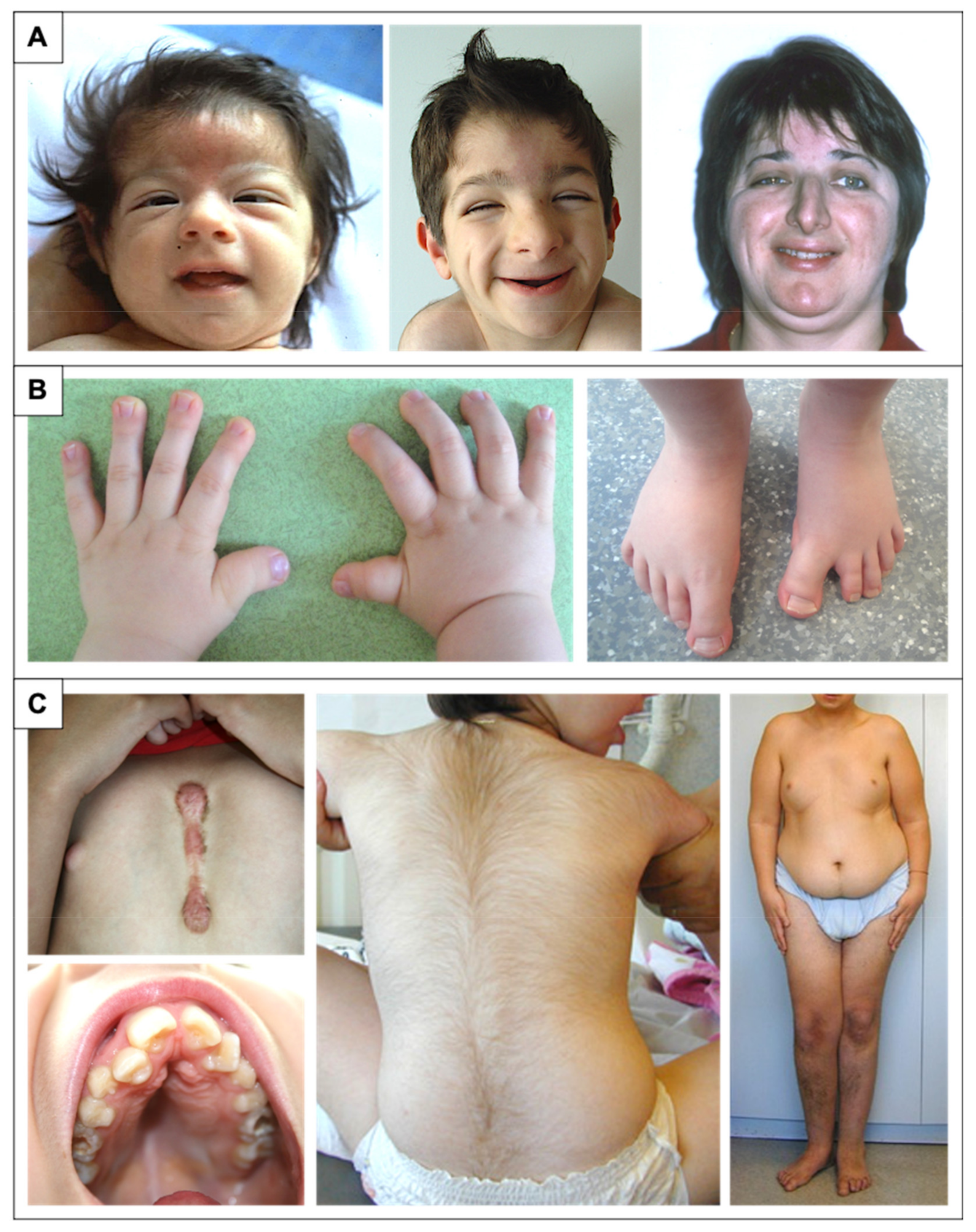
2.2. Facial Dysmorphism
2.3. Distal Limb and Skeletal Abnormalities
2.4. Development and Behavior
2.5. Growth Retardation and Microcephaly
2.6. Additional Features
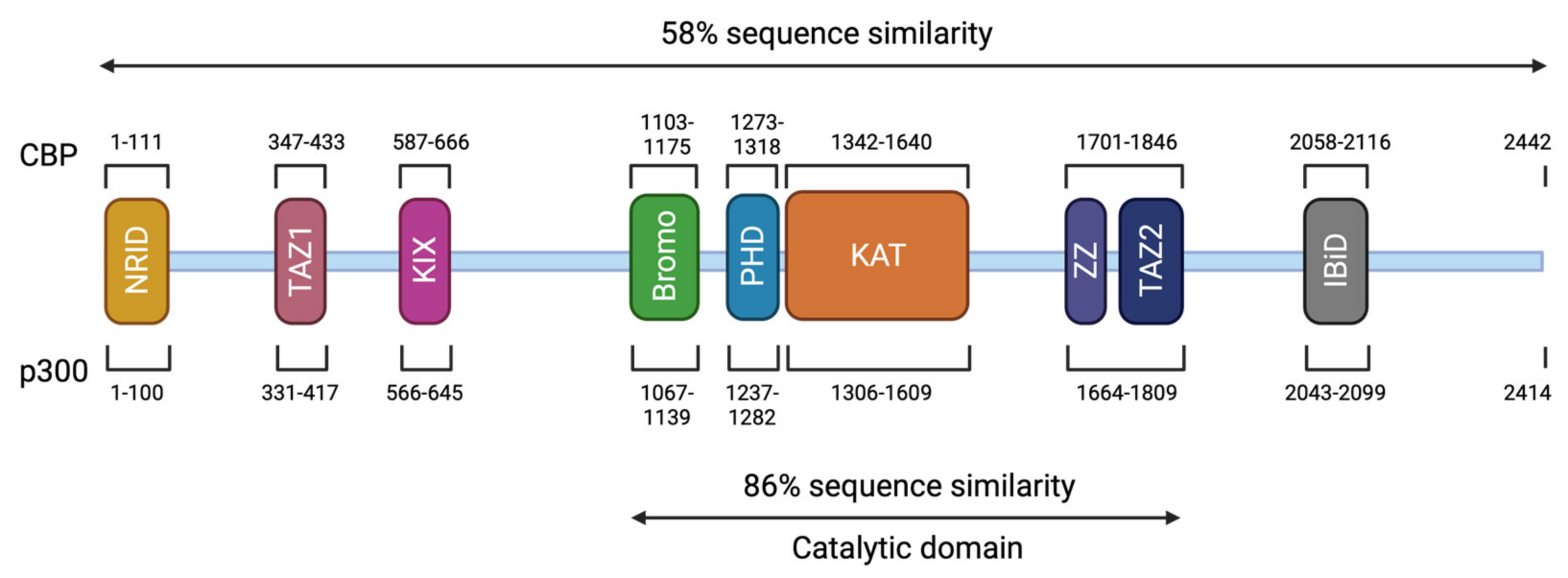
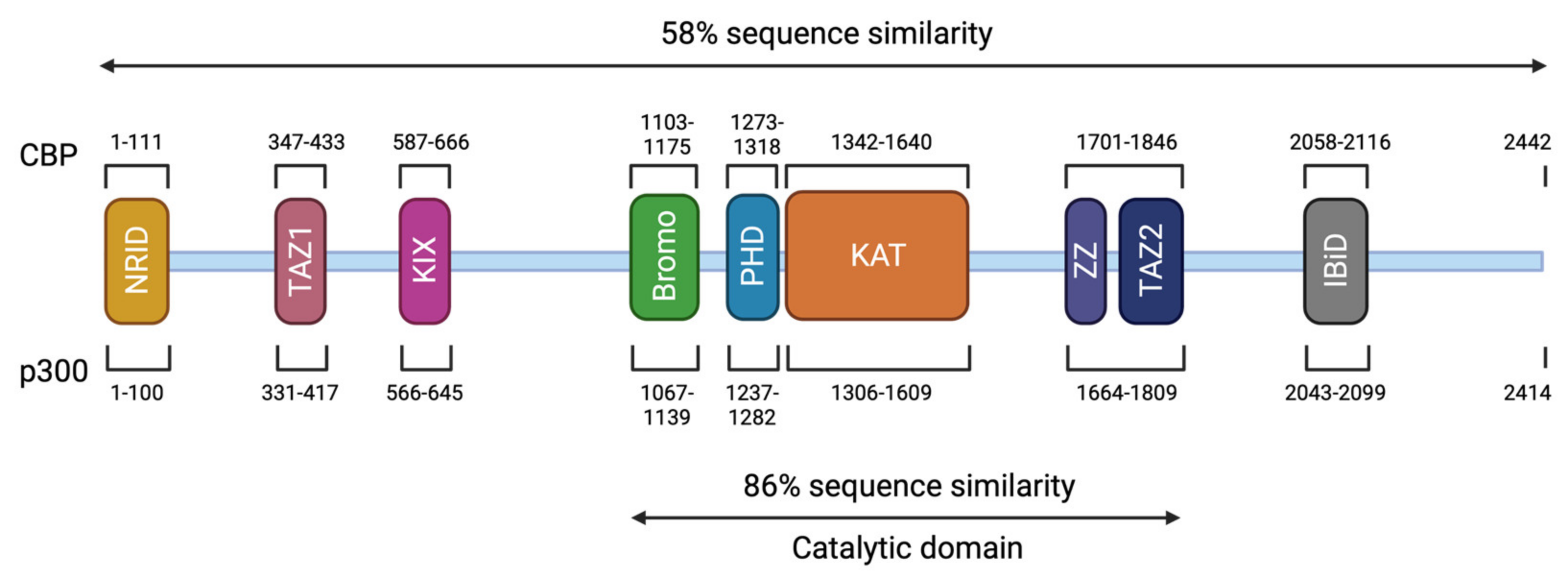
3. Genotype and Mutation Spectrum
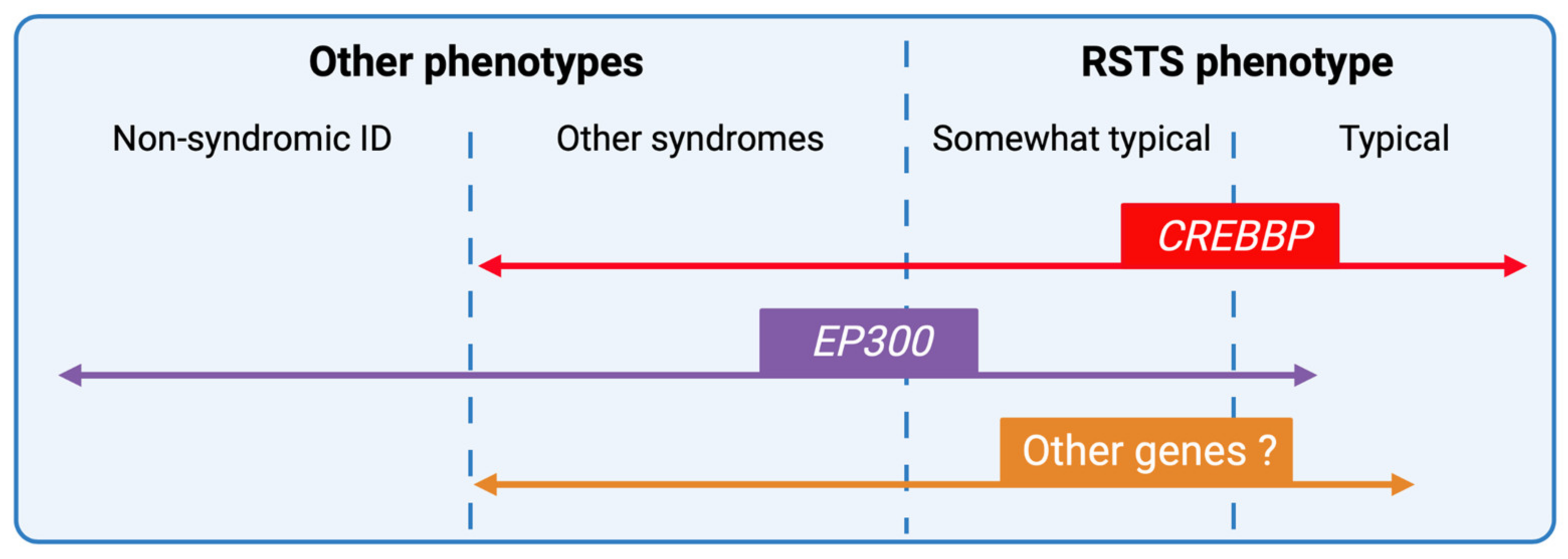
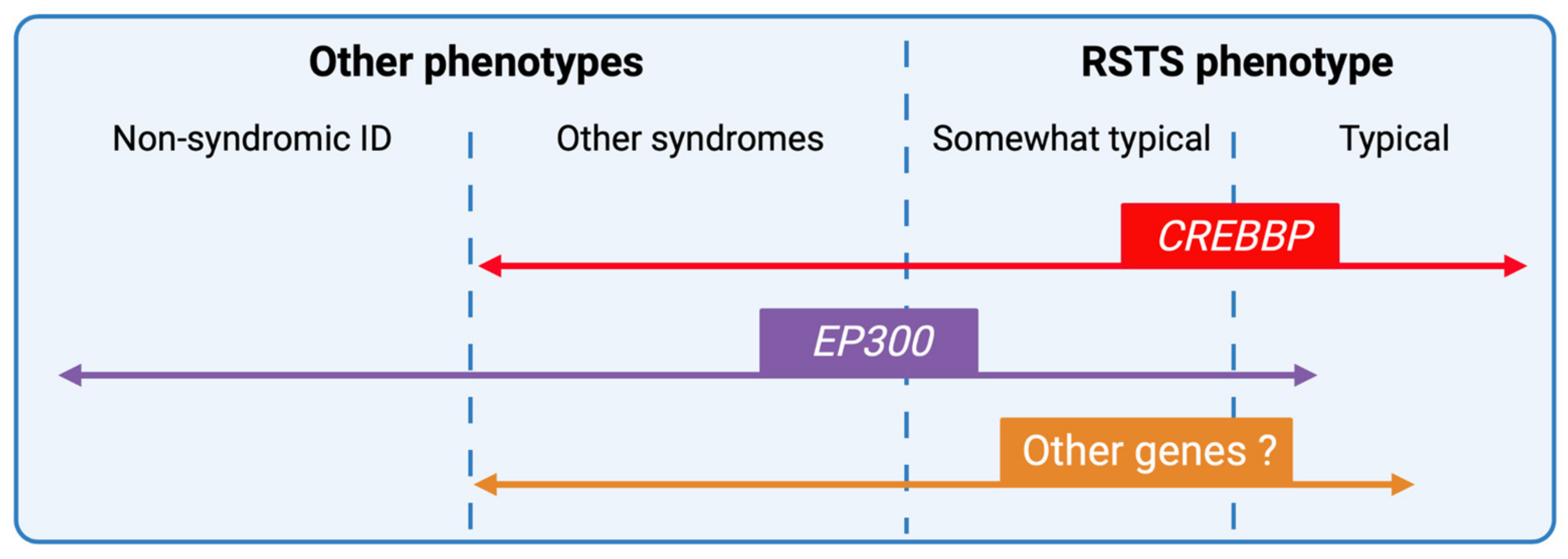
4. Phenotype-Genotype Correlations
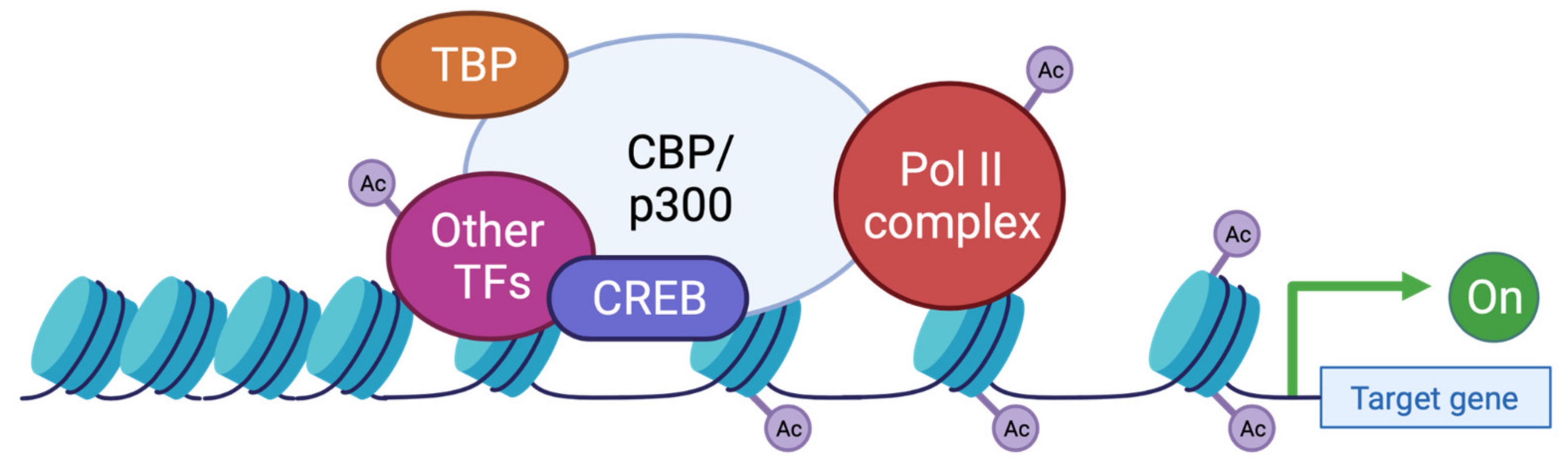
5. Epigenetic Regulation and Cognitive Function in RSTS
5.1. Syndromic Manifestations in the Mouse Model
5.2. Histone Acetylation Modifications and Memory Development
5.3. Role of KAT3 Proteins in Neurodevelopment and Cognitive Impairment
5.4. RSTS and Related Chromatinopathies
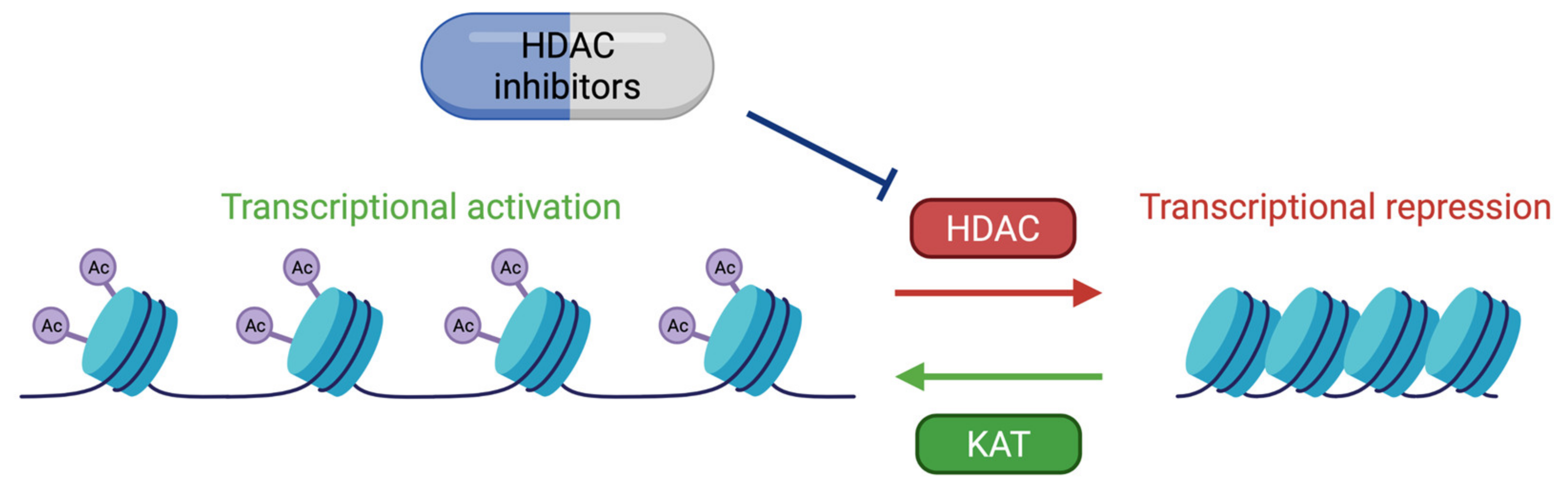
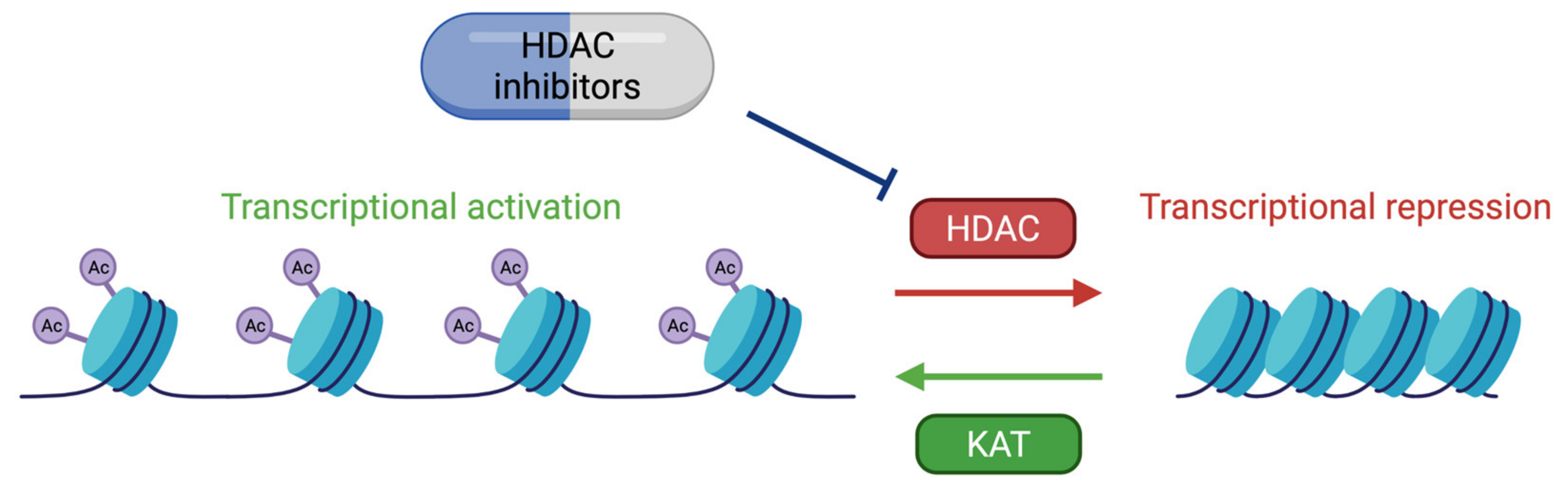
6. Therapeutic Approaches
7. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hennekam, R.C.M. Rubinstein–Taybi Syndrome. Eur. J. Hum. Genet. 2006, 14, 981–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, O.; Kress, W.; Kempf, O.; Lechno, S.; Haaf, T.; Zechner, U. Inheritance and Variable Expression in Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2010, 152A, 2254–2261. [Google Scholar] [CrossRef] [PubMed]
- López, M.; Seidel, V.; Santibáñez, P.; Cervera-Acedo, C.; Castro, P.C.; Domínguez-Garrido, E. First Case Report of Inherited Rubinstein-Taybi Syndrome Associated with a Novel EP300 Variant. BMC Med. Genet. 2016, 17, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennekam, R.C.; Lommen, E.J.; Strengers, J.L.; van Spijker, H.G.; Jansen-Kokx, T.M. Rubinstein-Taybi Syndrome in a Mother and Son. Eur. J. Pediatr. 1989, 148, 439–441. [Google Scholar] [CrossRef]
- Rubinstein, J.H. Broad Thumb-Hallux (Rubinstein-Taybi) Syndrome 1957–1988. Am. J. Med. Genet. 1990, 37, 3–16. [Google Scholar] [CrossRef]
- Rubinstein, J.H.; Taybi, H. Broad Thumbs and Toes and Facial Abnormalities. A Possible Mental Retardation Syndrome. Am. J. Dis. Child. 1963, 105, 588–608. [Google Scholar] [CrossRef]
- Hennekam, R.C.M.; van den Boogaard, M.-J.; Sibbles, B.J.; van Spijker, H.G. Rubinstein-Taybi Syndrome in the Netherlands. Am. J. Med. Genet. 1990, 37, 17–29. [Google Scholar] [CrossRef]
- Wiley, S.; Swayne, S.; Rubinstein, J.H.; Lanphear, N.E.; Stevens, C.A. Rubinstein-Taybi Syndrome Medical Guidelines. Am. J. Med. Genet. A 2003, 119A, 101–110. [Google Scholar] [CrossRef]
- Stevens, C.A.; Carey, J.C.; Blackburn, B.L. Rubinstein-Taybi Syndrome: A Natural History Study. Am. J. Med. Genet. A 1990, 37, 30–37. [Google Scholar] [CrossRef]
- Milani, D.; Manzoni, F.; Pezzani, L.; Ajmone, P.; Gervasini, C.; Menni, F.; Esposito, S. Rubinstein-Taybi Syndrome: Clinical Features, Genetic Basis, Diagnosis, and Management. Ital. J. Pediatr. 2015, 41, 4. [Google Scholar] [CrossRef] [Green Version]
- Boot, M.V.; van Belzen, M.J.; Overbeek, L.I.; Hijmering, N.; Mendeville, M.; Waisfisz, Q.; Wesseling, P.; Hennekam, R.C.; de Jong, D. Benign and Malignant Tumors in Rubinstein-Taybi Syndrome. Am. J. Med. Genet. A 2018, 176, 597–608. [Google Scholar] [CrossRef]
- Petrif, F.; Giles, R.H.; Dauwerse, H.G.; Saris, J.J.; Hennekam, R.C.M.; Masuno, M.; Tommerup, N.; van Ommen, G.-J.B.; Goodman, R.H.; Peters, D.J.M.; et al. Rubinstein-Taybi Syndrome Caused by Mutations in the Transcriptional Co-Activator CBP. Nature 1995, 376, 348–351. [Google Scholar] [CrossRef]
- Roelfsema, J.H.; White, S.J.; Ariyürek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; den Dunnen, J.T.; van Ommen, G.-J.B.; Breuning, M.H.; et al. Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Atalaya, J.P.; Valor, L.M.; Barco, A. Chapter—Epigenetic Factors in Intellectual Disability: The Rubinstein–Taybi Syndrome as a Paradigm of Neurodevelopmental Disorder with Epigenetic Origin. In Progress in Molecular Biology and Translational Science; Epigenetics and Neuroplasticity—Evidence and Debate; Lubin, F., Akbarian, S., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 128, pp. 139–176. [Google Scholar]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Postnatal Malleability and Therapeutic Prospects. Hum. Mol. Genet. 2019, 28, R254–R264. [Google Scholar] [CrossRef]
- Michail, J.; Matsoukas, J.; Theodorou, S. Arched, clubbed thumb in strong abduction-extension & other concomitant symptoms. Rev. Chir. Orthopédique Réparatrice Appar. Mot. 1957, 43, 142–146. [Google Scholar]
- Fergelot, P.; van Belzen, M.; van Gils, J.; Afenjar, A.; Armour, C.M.; Arveiler, B.; Beets, L.; Burglen, L.; Busa, T.; Collet, M.; et al. Phenotype and Genotype in 52 Patients with Rubinstein–Taybi Syndrome Caused by EP300 Mutations. Am. J. Med. Genet. A 2016, 170, 3069–3082. [Google Scholar] [CrossRef]
- Bartholdi, D.; Roelfsema, J.H.; Papadia, F.; Breuning, M.H.; Niedrist, D.; Hennekam, R.C.; Schinzel, A.; Peters, D.J.M. Genetic Heterogeneity in Rubinstein–Taybi Syndrome: Delineation of the Phenotype of the First Patients Carrying Mutations in EP300. J. Med. Genet. 2007, 44, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Foley, P.; Bunyan, D.; Stratton, J.; Dillon, M.; Lynch, S.A. Further Case of Rubinstein–Taybi Syndrome Due to a Deletion in EP300. Am. J. Med. Genet. A 2009, 149A, 997–1000. [Google Scholar] [CrossRef]
- Negri, G.; Milani, D.; Colapietro, P.; Forzano, F.; Della Monica, M.; Rusconi, D.; Consonni, L.; Caffi, L.G.; Finelli, P.; Scarano, G.; et al. Clinical and Molecular Characterization of Rubinstein-Taybi Syndrome Patients Carrying Distinct Novel Mutations of the EP300 Gene. Clin. Genet. 2015, 87, 148–154. [Google Scholar] [CrossRef]
- Cohen, J.L.; Schrier Vergano, S.A.; Mazzola, S.; Strong, A.; Keena, B.; McDougall, C.; Ritter, A.; Li, D.; Bedoukian, E.C.; Burke, L.W.; et al. EP300-Related Rubinstein-Taybi Syndrome: Highlighted Rare Phenotypic Findings and a Genotype-Phenotype Meta-Analysis of 74 Patients. Am. J. Med. Genet. A 2020, 182, 2926–2938. [Google Scholar] [CrossRef]
- Negri, G.; Magini, P.; Milani, D.; Colapietro, P.; Rusconi, D.; Scarano, E.; Bonati, M.T.; Priolo, M.; Crippa, M.; Mazzanti, L.; et al. From Whole Gene Deletion to Point Mutations of EP300-Positive Rubinstein–Taybi Patients: New Insights into the Mutational Spectrum and Peculiar Clinical Hallmarks. Hum. Mutat. 2016, 37, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-Eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Van-Gils, J.; Naudion, S.; Toutain, J.; Lancelot, G.; Attié-Bitach, T.; Blesson, S.; Demeer, B.; Doray, B.; Gonzales, M.; Martinovic, J.; et al. Fetal Phenotype of Rubinstein-Taybi Syndrome Caused by CREBBP Mutations. Clin. Genet. 2019, 95, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Cardalliac, C.; Vincent, M.; Joubert, M.; Vaillant, C.L. Rubinstein-Taybi Syndrome in a Fetus: Contribution of 2- and 3-Dimensional Ultrasonography. J. Ultrasound Med. 2018, 37, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.; Sglavo, G.; Paladini, D. Prenatal Sonographic Diagnosis of Rubinstein-Taybi Syndrome. J. Ultrasound Med. 2009, 28, 669–672. [Google Scholar] [CrossRef]
- Bedeschi, M.F.; Crippa, B.L.; Colombo, L.; Guez, S.; Cerruti, M.; Fogliani, R.; Gervasini, C.; Lalatta, F. Unusual Prenatal Presentation of Rubinstein–Taybi Syndrome: A Case Report. Am. J. Med. Genet. A 2014, 164, 2663–2666. [Google Scholar] [CrossRef]
- Simeonova-Brachot, I.I.; Gerony-Laffitte, L. Early Antenatal Sonographic Findings of Rubinstein-Taybi Syndrome: Imaging of High-Arched Palate and Bilateral Abducted Thumbs on Surface Rendering Mode at 17 Weeks. Ultrasound Int. Open 2018, 4, E139–E141. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosi, F.; Ronzoni, L.; Villa, R.; de Marinis, S.; Cetera, G.E.; Soldavini, C.M.; Ferrazzi, E. Ultrasound 2-D and 3-D Diagnosis of Rubinstein-Taybi Syndrome in a 21-Week-Old Fetus. J. Ultrasound 2020. [Google Scholar] [CrossRef]
- Tekendo-Ngongang, C.; Owosela, B.; Fleischer, N.; Addissie, Y.A.; Malonga, B.; Badoe, E.; Gupta, N.; Moresco, A.; Huckstadt, V.; Ashaat, E.A.; et al. Rubinstein-Taybi Syndrome in Diverse Populations. Am. J. Med. Genet. A 2020, 182, 2939–2950. [Google Scholar] [CrossRef]
- Beets, L.; Rodríguez-Fonseca, C.; Hennekam, R.C. Growth Charts for Individuals with Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2014, 164, 2300–2309. [Google Scholar] [CrossRef]
- Allanson, J.E. Rubinstein-Taybi Syndrome: The Changing Face. Am. J. Med. Genet. 1990, 37, 38–41. [Google Scholar] [CrossRef]
- Allanson, J.E. Microcephaly in Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 1993, 46, 244–246. [Google Scholar] [CrossRef]
- Stevens, C.A.; Hennekam, R.C.M.; Blackburn, B.L. Growth in the Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 1990, 37, 51–55. [Google Scholar] [CrossRef]
- Stevens, C.A.; Pouncey, J.; Knowles, D. Adults with Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2011, 155, 1680–1684. [Google Scholar] [CrossRef]
- Allanson, J.E.; Hennekam, R.C. Rubinstein-Taybi Syndrome: Objective Evaluation of Craniofacial Structure. Am. J. Med. Genet. A 1997, 71, 414–419. [Google Scholar] [CrossRef]
- Pérez-Grijalba, V.; García-Oguiza, A.; López, M.; Armstrong, J.; García-Miñaur, S.; Mesa-Latorre, J.M.; O’Callaghan, M.; Pineda Marfa, M.; Ramos-Arroyo, M.A.; Santos-Simarro, F.; et al. New Insights into Genetic Variant Spectrum and Genotype-Phenotype Correlations of Rubinstein-Taybi Syndrome in 39 CREBBP-Positive Patients. Mol. Genet. Genomic Med. 2019, 7, e972. [Google Scholar] [CrossRef]
- Schorry, E.K.; Keddache, M.; Lanphear, N.; Rubinstein, J.H.; Srodulski, S.; Fletcher, D.; Blough-Pfau, R.I.; Grabowski, G.A. Genotype–Phenotype Correlations in Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2008, 146A, 2512–2519. [Google Scholar] [CrossRef]
- Hutchinson, D.T.; Sullivan, R. Rubinstein-Taybi Syndrome. J. Hand Surg. 2015, 40, 1711–1712. [Google Scholar] [CrossRef]
- Hennekam, R.C.M.; van den Boogaard, M.-J.; Dijkstra, P.F.; van de Kamp, J.J.P. Metacarpophalangeal Pattern Profile Analysis in Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 1990, 37, 48–50. [Google Scholar] [CrossRef]
- Yu, S.; Wu, B.; Qian, Y.; Zhang, P.; Lu, Y.; Dong, X.; Wang, Q.; Zhao, X.; Liu, R.; Zhou, W.; et al. Clinical Exome Sequencing Identifies Novel CREBBP Variants in 18 Chinese Rubinstein-Taybi Syndrome Kids with High Frequency of Polydactyly. Mol. Genet. Genom. Med. 2019, 7, e1009. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kurosawa, K.; Masuno, M.; Okuzumi, S.; Kondo, S.; Miyama, S.; Okamoto, N.; Aida, N.; Nishimura, G. Congenital Anomaly of Cervical Vertebrae Is a Major Complication of Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2005, 135A, 130–133. [Google Scholar] [CrossRef]
- Ajmone, P.F.; Avignone, S.; Gervasini, C.; Giacobbe, A.; Monti, F.; Costantino, A.; Esposito, S.; Marchisio, P.; Triulzi, F.; Milani, D. Rubinstein–Taybi Syndrome: New Neuroradiological and Neuropsychiatric Insights from a Multidisciplinary Approach. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 406–415. [Google Scholar] [CrossRef]
- López, M.; García-Oguiza, A.; Armstrong, J.; García-Cobaleda, I.; García-Miñaur, S.; Santos-Simarro, F.; Seidel, V.; Domínguez-Garrido, E. Rubinstein-Taybi 2 Associated to Novel EP300 Mutations: Deepening the Clinical and Genetic Spectrum. BMC Med. Genet. 2018, 19, 36. [Google Scholar] [CrossRef] [Green Version]
- Crawford, H.; Moss, J.; Groves, L.; Dowlen, R.; Nelson, L.; Reid, D.; Oliver, C. A Behavioural Assessment of Social Anxiety and Social Motivation in Fragile X, Cornelia de Lange and Rubinstein-Taybi Syndromes. J. Autism Dev. Disord. 2020, 50, 127–144. [Google Scholar] [CrossRef]
- Hennekam, R.C.; Baselier, A.C.; Beyaert, E.; Bos, A.; Blok, J.B.; Jansma, H.B.; Thorbecke-Nilsen, V.V.; Veerman, H. Psychological and Speech Studies in Rubinstein-Taybi Syndrome. Am. J. Ment. Retard. AJMR 1992, 96, 645–660. [Google Scholar]
- Waite, J.; Moss, J.; Beck, S.R.; Richards, C.; Nelson, L.; Arron, K.; Burbidge, C.; Berg, K.; Oliver, C. Repetitive Behavior in Rubinstein–Taybi Syndrome: Parallels with Autism Spectrum Phenomenology. J. Autism Dev. Disord. 2015, 45, 1238–1253. [Google Scholar] [CrossRef] [Green Version]
- Cazalets, J.R.; Bestaven, E.; Doat, E.; Baudier, M.P.; Gallot, C.; Amestoy, A.; Bouvard, M.; Guillaud, E.; Guillain, I.; Grech, E.; et al. Evaluation of Motor Skills in Children with Rubinstein–Taybi Syndrome. J. Autism Dev. Disord. 2017, 47, 3321–3332. [Google Scholar] [CrossRef]
- Galéra, C.; Taupiac, E.; Fraisse, S.; Naudion, S.; Toussaint, E.; Rooryck-Thambo, C.; Delrue, M.-A.; Arveiler, B.; Lacombe, D.; Bouvard, M.-P. Socio-Behavioral Characteristics of Children with Rubinstein-Taybi Syndrome. J. Autism Dev. Disord. 2009, 39, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Levitas, A.S.; Reid, C.S. Rubinstein-Taybi Syndrome and Psychiatric Disorders. J. Intellect. Disabil. Res. 1998, 42, 284–292. [Google Scholar] [CrossRef]
- Yagihashi, T.; Kosaki, K.; Okamoto, N.; Mizuno, S.; Kurosawa, K.; Takahashi, T.; Sato, Y.; Kosaki, R. Age-Dependent Change in Behavioral Feature in Rubinstein-Taybi Syndrome. Congenit. Anom. 2012, 52, 82–86. [Google Scholar] [CrossRef]
- Taupiac, E.; Lacombe, D.; Thiébaut, E.; Van-Gils, J.; Michel, G.; Fergelot, P.; Adrien, J.-L. Psychomotor, Cognitive, and Socio-Emotional Developmental Profiles of Children with Rubinstein-Taybi Syndrome and a Severe Intellectual Disability. J. Intellect. Dev. Disabil. 2020, 0, 1–10. [Google Scholar] [CrossRef]
- Giacobbe, A.; Ajmone, P.F.; Milani, D.; Avignone, S.; Triulzi, F.; Gervasini, C.; Menni, F.; Monti, F.; Biffi, D.; Canavesi, K.; et al. Electroclinical Phenotype in Rubinstein-Taybi Syndrome. Brain Dev. 2016, 38, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Cantani, A.; Gagliesi, D. Rubinstein-Taybi Syndrome. Review of 732 Cases and Analysis of the Typical Traits. Eur. Rev. Med. Pharmacol. Sci. 1998, 2, 81–87. [Google Scholar]
- Mishra, S.; Agarwalla, S.K.; Potpalle, D.R.; Dash, N.N. Rubinstein-Taybi Syndrome with Agenesis of Corpus Callosum. J. Pediatr. Neurosci. 2015, 10, 175–177. [Google Scholar] [CrossRef]
- Hadzsiev, K.; Gyorsok, Z.; Till, A.; Czakó, M.; Bartsch, O. Rubinstein–Taybi Syndrome 2 with Cerebellar Abnormality and Neural Tube Defect. Clin. Dysmorphol. 2019, 28, 135–139. [Google Scholar] [CrossRef]
- Guion-Almeida, M.L.; Richieri-Costa, A. Callosal Agenesis, Iris Coloboma, and Megacolon in a Brazilian Boy with Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 1992, 43, 929–931. [Google Scholar] [CrossRef]
- Marzuillo, P.; Grandone, A.; Coppola, R.; Cozzolino, D.; Festa, A.; Messa, F.; Luongo, C.; Del Giudice, E.M.; Perrone, L. Novel CAMP Binding Protein-BP (CREBBP) Mutation in a Girl with Rubinstein-Taybi Syndrome, GH Deficiency, Arnold Chiari Malformation and Pituitary Hypoplasia. BMC Med. Genet. 2013, 14, 28. [Google Scholar] [CrossRef]
- Lee, J.S.; Byun, C.K.; Kim, H.; Lim, B.C.; Hwang, H.; Choi, J.E.; Hwang, Y.S.; Seong, M.-W.; Park, S.S.; Kim, K.J.; et al. Clinical and Mutational Spectrum in Korean Patients with Rubinstein–Taybi Syndrome: The Spectrum of Brain MRI Abnormalities. Brain Dev. 2015, 37, 402–408. [Google Scholar] [CrossRef]
- Tanaka, T.; Ling, B.C.; Rubinstein, J.H.; Crone, K.R. Rubinstein–Taybi Syndrome in Children with Tethered Spinal Cord. J. Neurosurg. Pediatr. 2006, 105, 261–264. [Google Scholar] [CrossRef]
- Stevens, C.A.; Bhakta, M.G. Cardiac Abnormalities in the Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 1995, 59, 346–348. [Google Scholar] [CrossRef]
- Loomba, R.S.; Geddes, G. Tricuspid Atresia and Pulmonary Atresia in a Child with Rubinstein-Taybi Syndrome. Ann. Pediatr. Cardiol. 2015, 8, 157. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Stachtou, J.; Emmanouil, D.; Arveiler, B.; Griffiths, D.; Lacombe, D. Oro-Dental Features as Useful Diagnostic Tool in Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2007, 143A, 570–573. [Google Scholar] [CrossRef]
- Sharma, N.; Mali, A.M.; Bapat, S.A. Spectrum of CREBBP Mutations in Indian Patients with Rubinstein–Taybi Syndrome. J. Biosci. 2010, 35, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.M.; Naik, M.N.; Ali, M.J. Lacrimal Drainage Anomalies in Rubinstein–Taybi Syndrome: Case Report and Review of Literature. Orbit 2019, 38, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Van Genderen, M.M.; Kinds, G.; Riemslag, F.; Hennekam, R. Ocular Features in Rubinstein–Taybi Syndrome: Investigation of 24 Patients and Review of the Literature. Br. J. Ophthalmol. 2000, 84, 1177–1184. [Google Scholar] [CrossRef]
- Brei, T.J.; Burke, M.J.; Rubinstein, J.H. Glaucoma and Findings Simulating Glaucoma in the Rubinstein-Taybi Syndrome. J. Pediatr. Ophthalmol. Strabismus 1995, 32, 248–252. [Google Scholar] [CrossRef]
- Grunow, J.E. Gastroesophageal Reflux in Rubinstein--Taybi Syndrome. J. Pediatr. Gastroenterol. Nutr. 1982, 1, 273–274. [Google Scholar] [CrossRef]
- Isidor, B.; Podevin, G.; Camby, C.; Mosnier, J.-F.; Chauty, A.; Lyet, J.-M.; Fergelot, P.; Lacombe, D.; Arveiler, B.; Pelet, A.; et al. Rubinstein–Taybi Syndrome and Hirschsprung Disease in a Patient Harboring an Intragenic Deletion of the CREBBP Gene. Am. J. Med. Genet. A 2010, 152A, 1847–1848. [Google Scholar] [CrossRef]
- Lee, K.H.; Park, E.Y.; Jung, S.W.; Song, S.W.; Lim, H.K. Hysterectomy Due to Abnormal Uterine Bleeding in a 15-Year Old Girl with Rubinstein-Taybi Syndrome. J. Lifestyle Med. 2016, 6, 76–78. [Google Scholar] [CrossRef] [Green Version]
- De Castro Coelho, F.; Câmara, S.; Alves, I.; Brazão, K. Septate Uterus in a Girl with Rubinstein–Taybi Syndrome. Available online: https://www.hindawi.com/journals/cripe/2018/7878156/ (accessed on 10 December 2020).
- Kuwabara, J.; Akita, S.; Sato, M.; Watanabe, K.; Tanigawa, K.; Matsuno, Y.; Abe, Y.; Kikuchi, S.; Yoshida, M.; Koga, S.; et al. Paraovarian Cyst Torsion in a Patient with Rubinstein–Taybi Syndrome: A Case Report. J. Nippon Med. Sch. 2020, advpub. [Google Scholar] [CrossRef]
- Van de Kar, A.L.; Houge, G.; Shaw, A.C.; de Jong, D.; van Belzen, M.J.; Peters, D.J.M.; Hennekam, R.C.M. Keloids in Rubinstein–Taybi Syndrome: A Clinical Study. Br. J. Dermatol. 2014, 171, 615–621. [Google Scholar] [CrossRef]
- Saettini, F.; Herriot, R.; Prada, E.; Nizon, M.; Zama, D.; Marzollo, A.; Romaniouk, I.; Lougaris, V.; Cortesi, M.; Morreale, A.; et al. Prevalence of Immunological Defects in a Cohort of 97 Rubinstein-Taybi Syndrome Patients. J. Clin. Immunol. 2020, 40, 851–860. [Google Scholar] [CrossRef]
- Miller, R.W.; Rubinstein, J.H. Tumors in Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 1995, 56, 112–115. [Google Scholar] [CrossRef]
- Siraganian, P.A.; Rubinstein, J.H.; Miller, R.W. Keloids and Neoplasms in the Rubinstein-Taybi Syndrome. Med. Pediatr. Oncol. 1989, 17, 485–491. [Google Scholar] [CrossRef]
- Milani, D.; Bonarrigo, F.A.; Menni, F.; Spaccini, L.; Gervasini, C.; Esposito, S. Hepatoblastoma in Rubinstein–Taybi Syndrome: A Case Report. Pediatr. Blood Cancer 2016, 63, 572–573. [Google Scholar] [CrossRef]
- Rokunohe, D.; Nakano, H.; Akasaka, E.; Toyomaki, Y.; Sawamura, D. Rubinstein-Taybi Syndrome with Multiple Pilomatricomas: The First Case Diagnosed by CREBBP Mutation Analysis. J. Dermatol. Sci. 2016, 83, 240–242. [Google Scholar] [CrossRef]
- Bueno, A.L.A.; de Souza, M.E.V.; Graziadio, C.; Kiszewski, A.E. Multiple Pilomatricomas in Twins with Rubinstein–Taybi Syndrome. An. Bras. Dermatol. 2020, 95, 619–622. [Google Scholar] [CrossRef]
- Sy, C.; Henry, J.; Kura, B.; Brenner, A.; Grandhi, R. Primary Diffuse Large B-Cell Lymphoma in a Patient with Rubinstein-Taybi Syndrome: Case Report and Review of the Literature. World Neurosurg. 2018, 109, 342–346. [Google Scholar] [CrossRef]
- Lacombe, D.; Saura, R.; Taine, L.; Battin, J. Confirmation of Assigment of a Locus for Rubinstein-Taybi Syndrome Gene to 16p13.3. Am. J. Med. Genet. 1992, 44, 126–128. [Google Scholar] [CrossRef]
- Chiang, P.-W.; Lee, N.-C.; Chien, N.; Hwu, W.-L.; Spector, E.; Tsai, A.C.-H. Somatic and Germ-Line Mosaicism in Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2009, 149A, 1463–1467. [Google Scholar] [CrossRef]
- De Vries, T.I.; Monroe, G.R.; van Belzen, M.J.; van der Lans, C.A.; Savelberg, S.M.; Newman, W.G.; van Haaften, G.; Nievelstein, R.A.; van Haelst, M.M. Mosaic CREBBP Mutation Causes Overlapping Clinical Features of Rubinstein-Taybi and Filippi Syndromes. Eur. J. Hum. Genet. EJHG 2016, 24, 1363–1366. [Google Scholar] [CrossRef] [Green Version]
- Gucev, Z.S.; Tasic, V.B.; Saveski, A.; Polenakovic, M.H.; Laban, N.B.; Zechner, U.; Bartsch, O. Tissue-Specific Mosaicism in a Patient with Rubinstein–Taybi Syndrome and CREBBP Exon 1 Duplication. Clin. Dysmorphol. 2019, 28, 140–142. [Google Scholar] [CrossRef]
- Cotsirilos, P.; Taylor, J.C.; Matalon, R. Dominant Inheritance of a Syndrome Similar to Rubinstein-Taybi. Am. J. Med. Genet. 1987, 26, 85–93. [Google Scholar] [CrossRef]
- Marion, R.W.; Garcia, D.M.; Karasik, J.B. Apparent Dominant Transmission of the Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 1993, 46, 284–287. [Google Scholar] [CrossRef]
- Petrij, F.; Dauwerse, H.G.; Blough, R.I.; Giles, R.H.; van der Smagt, J.J.; Wallerstein, R.; Maaswinkel-Mooy, P.D.; van Karnebeek, C.D.; van Ommen, G.-J.B.; van Haeringen, A.; et al. Diagnostic Analysis of the Rubinstein-Taybi Syndrome: Five Cosmids Should Be Used for Microdeletion Detection and Low Number of Protein Truncating Mutations. J. Med. Genet. 2000, 37, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, K.; Kuroki, Y. Rubinstein-Taybi Syndrome with de Novo Reciprocal Translocation t(2;16) (P13.3; P13.3). Am. J. Med. Genet. 1991, 38, 636–639. [Google Scholar] [CrossRef]
- Imaizumi, K.; Kurosawa, K.; Masuno, M.; Tsukahara, M.; Kuroki, Y. Chromosome Aberrations in Rubinstein-Taybi Syndrome. Clin. Genet. 1993, 43, 215–216. [Google Scholar] [CrossRef]
- Tommerup, N.; van der Hagen, C.B.; Heiberg, A. Tentative Assignment of a Locus for Rubinstein-Taybi Syndrome to 16p13.3 by a de Novo Reciprocal Translocation, t(7;16)(Q34;P13.3). Am. J. Med. Genet. 1992, 44, 237–241. [Google Scholar] [CrossRef]
- Chrivia, J.C.; Kwok, R.P.S.; Lamb, N.; Hagiwara, M.; Montminy, M.R.; Goodman, R.H. Phosphorylated CREB Binds Specifically to the Nuclear Protein CBP. Nature 1993, 365, 855–859. [Google Scholar] [CrossRef]
- Whyte, P.; Williamson, N.M.; Harlow, E. Cellular Targets for Transformation by the Adenovirus E1A Proteins. Cell 1989, 56, 67–75. [Google Scholar] [CrossRef]
- Egan, C.; Jelsma, T.N.; Howe, J.A.; Bayley, S.T.; Ferguson, B.; Branton, P.E. Mapping of Cellular Protein-Binding Sites on the Products of Early-Region 1A of Human Adenovirus Type 5. Mol. Cell. Biol. 1988, 8, 3955–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, O.; Schmidt, S.; Richter, M.; Morlot, S.; Seemanová, E.; Wiebe, G.; Rasi, S. DNA Sequencing of CREBBP Demonstrates Mutations in 56% of Patients with Rubinstein–Taybi Syndrome (RSTS) and in Another Patient with Incomplete RSTS. Hum. Genet. 2005, 117, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Kalkhoven, E.; Roelfsema, J.H.; Teunissen, H.; den Boer, A.; Ariyurek, Y.; Zantema, A.; Breuning, M.H.; Hennekam, R.C.M.; Peters, D.J.M. Loss of CBP Acetyltransferase Activity by PHD Finger Mutations in Rubinstein–Taybi Syndrome. Hum. Mol. Genet. 2003, 12, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Coupry, I.; Roudaut, C.; Stef, M.; Delrue, M.-A.; Marche, M.; Burgelin, I.; Taine, L.; Cruaud, C.; Lacombe, D.; Arveiler, B. Molecular Analysis of the CBP Gene in 60 Patients with Rubinstein-Taybi Syndrome. J. Med. Genet. 2002, 39, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Kurokawa, R.; Krones, A.; Tatsumi, K.; Ishii, M.; Taki, T.; Masuno, M.; Ohashi, H.; Yanagisawa, M.; Rosenfeld, M.G.; et al. Defect of Histone Acetyltransferase Activity of the Nuclear Transcriptional Coactivator CBP in Rubinstein–Taybi Syndrome. Hum. Mol. Genet. 2001, 10, 1071–1076. [Google Scholar] [CrossRef] [Green Version]
- Udaka, T.; Samejima, H.; Kosaki, R.; Kurosawa, K.; Okamoto, N.; Mizuno, S.; Makita, Y.; Numabe, H.; Toral, J.F.; Takahashi, T.; et al. Comprehensive Screening of CREB-Binding Protein Gene Mutations among Patients with Rubinstein-Taybi Syndrome Using Denaturing High-Performance Liquid Chromatography. Congenit. Anom. 2005, 45, 125–131. [Google Scholar] [CrossRef]
- Cross, E.; Duncan-Flavell, P.J.; Howarth, R.J.; Hobbs, J.I.; Thomas, N.S.; Bunyan, D.J. Screening of a Large Rubinstein-Taybi Cohort Identified Many Novel Variants and Emphasizes the Importance of the CREBBP Histone Acetyltransferase Domain. Am. J. Med. Genet. A 2020, 182, 2508–2520. [Google Scholar] [CrossRef]
- Base HGMD. Available online: https://Portal.Biobase-International.Com/Hgmd/pro/Start.Php (accessed on 27 April 2021).
- Home—LOVD—An Open Source DNA Variation Database System. Available online: http://www.lovd.nl/3.0/home (accessed on 27 April 2021).
- Coupry, I.; Monnet, L.; Moneim Attia, A.A.E.; Taine, L.; Lacombe, D.; Arveiler, B. Analysis of CBP (CREBBP) Gene Deletions in Rubinstein-Taybi Syndrome Patients Using Real-Time Quantitative PCR. Hum. Mutat. 2004, 23, 278–284. [Google Scholar] [CrossRef]
- Gervasini, C.; Castronovo, P.; Bentivegna, A.; Mottadelli, F.; Faravelli, F.; Giovannucci-Uzielli, M.L.; Pessagno, A.; Lucci-Cordisco, E.; Pinto, A.M.; Salviati, L.; et al. High Frequency of Mosaic CREBBP Deletions in Rubinstein–Taybi Syndrome Patients and Mapping of Somatic and Germ-Line Breakpoints. Genomics 2007, 90, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Rusconi, D.; Negri, G.; Colapietro, P.; Picinelli, C.; Milani, D.; Spena, S.; Magnani, C.; Silengo, M.C.; Sorasio, L.; Curtisova, V.; et al. Characterization of 14 Novel Deletions Underlying Rubinstein–Taybi Syndrome: An Update of the CREBBP Deletion Repertoire. Hum. Genet. 2015, 134, 613–626. [Google Scholar] [CrossRef]
- Tsai, A.C.-H.; Dossett, C.J.; Walton, C.S.; Cramer, A.E.; Eng, P.A.; Nowakowska, B.A.; Pursley, A.N.; Stankiewicz, P.; Wiszniewska, J.; Cheung, S.W. Exon Deletions of the EP300 and CREBBP Genes in Two Children with Rubinstein–Taybi Syndrome Detected by ACGH. Eur. J. Hum. Genet. 2011, 19, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Wincent, J.; Luthman, A.; van Belzen, M.; van der Lans, C.; Albert, J.; Nordgren, A.; Anderlid, B.-M. CREBBP and EP300 Mutational Spectrum and Clinical Presentations in a Cohort of Swedish Patients with Rubinstein–Taybi Syndrome. Mol. Genet. Genom. Med. 2016, 4, 39–45. [Google Scholar] [CrossRef]
- Zimmermann, N.; Acosta, A.M.B.F.; Kohlhase, J.; Bartsch, O. Confirmation of EP300 Gene Mutations as a Rare Cause of Rubinstein–Taybi Syndrome. Eur. J. Hum. Genet. 2007, 15, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Giles, R.H.; Dauwerse, H.G.; van Ommen, G.J.; Breuning, M.H. Do Human Chromosomal Bands 16p13 and 22q11-13 Share Ancestral Origins? Am. J. Hum. Genet. 1998, 63, 1240–1242. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New Nomenclature for Chromatin-Modifying Enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Dancy, B.M.; Cole, P.A. Protein Lysine Acetylation by P300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.E.; Mapp, A.K. Modulating the Masters: Chemical Tools to Dissect CBP and P300 Function. Curr. Opin. Chem. Biol. 2018, 45, 195–203. [Google Scholar] [CrossRef]
- Valor, L.; Viosca, J.; Lopez-Atalaya, J.; Barco, A. Lysine Acetyltransferases CBP and P300 as Therapeutic Targets in Cognitive and Neurodegenerative Disorders. Curr. Pharm. Des. 2013, 19, 5051–5064. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-J.; Seto, E. Lysine Acetylation: Codified Crosstalk with Other Posttranslational Modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Bedford, D.C.; Kasper, L.H.; Fukuyama, T.; Brindle, P.K. Target Gene Context Influences the Transcriptional Requirement for the KAT3 Family of CBP and P300 Histone Acetyltransferases. Epigenetics Off. J. DNA Methylation Soc. 2010, 5, 9–15. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y.; Cole, P.A.; Marmorstein, R. Structure and Chemistry of the P300/CBP and Rtt109 Histone Acetyltransferases: Implications for Histone Acetyltransferase Evolution and Function. Curr. Opin. Struct. Biol. 2008, 18, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Histol Histopathol, Vol 17, Janknecht. Available online: http://www.hh.um.es/Abstracts/Vol_17/17_2/17_2_657.htm (accessed on 27 July 2016).
- Iyer, N.G.; Özdag, H.; Caldas, C. P300/CBP and Cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunstein, M. Histone Acetylation in Chromatin Structure and Transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-Wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Hasan, S.; Hassa, P.O.; Imhof, R.; Hottiger, M.O. Transcription Coactivator P300 Binds PCNA and May Have a Role in DNA Repair Synthesis. Nature 2001, 410, 387–391. [Google Scholar] [CrossRef]
- Tini, M.; Benecke, A.; Um, S.-J.; Torchia, J.; Evans, R.M.; Chambon, P. Association of CBP/P300 Acetylase and Thymine DNA Glycosylase Links DNA Repair and Transcription. Mol. Cell 2002, 9, 265–277. [Google Scholar] [CrossRef]
- Bannister, A.J.; Miska, E.A.; Görlich, D.; Kouzarides, T. Acetylation of Importin-α Nuclear Import Factors by CBP/P300. Curr. Biol. 2000, 10, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Spena, S.; Milani, D.; Rusconi, D.; Negri, G.; Colapietro, P.; Elcioglu, N.; Bedeschi, F.; Pilotta, A.; Spaccini, L.; Ficcadenti, A.; et al. Insights into Genotype–Phenotype Correlations from CREBBP Point Mutation Screening in a Cohort of 46 Rubinstein–Taybi Syndrome Patients. Clin. Genet. 2015, 88, 431–440. [Google Scholar] [CrossRef]
- Bentivegna, A.; Milani, D.; Gervasini, C.; Castronovo, P.; Mottadelli, F.; Manzini, S.; Colapietro, P.; Giordano, L.; Atzeri, F.; Divizia, M.T.; et al. Rubinstein-Taybi Syndrome: Spectrum of CREBBP Mutations in Italian Patients. BMC Med. Genet. 2006, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.D.; Bodian, D.L.; Khromykh, A.; Mora, G.G.; Lanpher, B.C.; Iyer, R.K.; Baveja, R.; Vockley, J.G.; Niederhuber, J.E. Expanding the Phenotypic Spectrum in EP300-Related Rubinstein–Taybi Syndrome. Am. J. Med. Genet. A 2015, 167, 1111–1116. [Google Scholar] [CrossRef]
- Yu, P.T.; Luk, H.-M.; Lo, I.F.M. Rubinstein-Taybi Syndrome in Chinese Population with Four Novel Mutations. Am. J. Med. Genet. A. 2020. [Google Scholar] [CrossRef]
- Bartsch, O.; Rasi, S.; Delicado, A.; Dyack, S.; Neumann, L.M.; Seemanová, E.; Volleth, M.; Haaf, T.; Kalscheuer, V.M. Evidence for a New Contiguous Gene Syndrome, the Chromosome 16p13.3 Deletion Syndrome Alias Severe Rubinstein–Taybi Syndrome. Hum. Genet. 2006, 120, 179–186. [Google Scholar] [CrossRef]
- Menke, L.A.; van Belzen, M.J.; Alders, M.; Cristofoli, F.; The DDD Study; Ehmke, N.; Fergelot, P.; Foster, A.; Gerkes, E.H.; Hoffer, M.J.V.; et al. CREBBP Mutations in Individuals without Rubinstein–Taybi Syndrome Phenotype. Am. J. Med. Genet. A. 2016. [Google Scholar] [CrossRef]
- Menke, L.A.; DDD Study; Gardeitchik, T.; Hammond, P.; Heimdal, K.R.; Houge, G.; Hufnagel, S.B.; Ji, J.; Johansson, S.; Kant, S.G.; et al. Further Delineation of an Entity Caused by CREBBP and EP300 Mutations but Not Resembling Rubinstein-Taybi Syndrome. Am. J. Med. Genet. A 2018, 176, 862–876. [Google Scholar] [CrossRef]
- Banka, S.; Sayer, R.; Breen, C.; Barton, S.; Pavaine, J.; Sheppard, S.E.; Bedoukian, E.; Skraban, C.; Cuddapah, V.A.; Clayton-Smith, J. Genotype-Phenotype Specificity in Menke-Hennekam Syndrome Caused by Missense Variants in Exon 30 or 31 of CREBBP. Am. J. Med. Genet. A 2019, 179, 1058–1062. [Google Scholar] [CrossRef] [Green Version]
- De Guzman, R.N.; Liu, H.Y.; Martinez-Yamout, M.; Dyson, H.J.; Wright, P.E. Solution Structure of the TAZ2 (CH3) Domain of the Transcriptional Adaptor Protein CBP1. J. Mol. Biol. 2000, 303, 243–253. [Google Scholar] [CrossRef]
- Ponting, C.P.; Blake, D.J.; Davies, K.E.; Kendrick-Jones, J.; Winder, S.J. ZZ and TAZ: New Putative Zinc Fingers in Dystrophin and Other Proteins. Trends Biochem. Sci. 1996, 21, 11–13. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Alari, V.; Russo, S.; Terragni, B.; Ajmone, P.F.; Sironi, A.; Catusi, I.; Calzari, L.; Concolino, D.; Marotta, R.; Milani, D.; et al. IPSC-Derived Neurons of CREBBP- and EP300-Mutated Rubinstein-Taybi Syndrome Patients Show Morphological Alterations and Hypoexcitability. Stem Cell Res. 2018, 30, 130–140. [Google Scholar] [CrossRef]
- Calzari, L.; Barcella, M.; Alari, V.; Braga, D.; Muñoz-Viana, R.; Barlassina, C.; Finelli, P.; Gervasini, C.; Barco, A.; Russo, S.; et al. Transcriptome Analysis of IPSC-Derived Neurons from Rubinstein-Taybi Patients Reveals Deficits in Neuronal Differentiation. Mol. Neurobiol. 2020, 57, 3685–3701. [Google Scholar] [CrossRef]
- Oike, Y.; Hata, A.; Mamiya, T.; Kaname, T.; Noda, Y.; Suzuki, M.; Yasue, H.; Nabeshima, T.; Araki, K.; Yamamura, K. Truncated CBP Protein Leads to Classical Rubinstein—Taybi Syndrome Phenotypes in Mice: Implications for a Dominant-Negative Mechanism. Hum. Mol. Genet. 1999, 8, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Kung, A.L.; Rebel, V.I.; Bronson, R.T.; Ch’ng, L.-E.; Sieff, C.A.; Livingston, D.M.; Yao, T.-P. Gene Dose-Dependent Control of Hematopoiesis and Hematologic Tumor Suppression by CBP. Genes Dev. 2000, 14, 272–277. [Google Scholar]
- Viosca, J.; Lopez-Atalaya, J.P.; Olivares, R.; Eckner, R.; Barco, A. Syndromic Features and Mild Cognitive Impairment in Mice with Genetic Reduction on P300 Activity: Differential Contribution of P300 and CBP to Rubinstein–Taybi Syndrome Etiology. Neurobiol. Dis. 2010, 37, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Naruse, I.; Hongo, T.; Xu, M.-J.; Nakahata, T.; Maekawa, T.; Ishii, S. Extensive Brain Hemorrhage and Embryonic Lethality in a Mouse Null Mutant of CREB-Binding Protein. Mech. Dev. 2000, 95, 133–145. [Google Scholar] [CrossRef]
- Yamauchi, T.; Oike, Y.; Kamon, J.; Waki, H.; Komeda, K.; Tsuchida, A.; Date, Y.; Li, M.-X.; Miki, H.; Akanuma, Y.; et al. Increased Insulin Sensitivity despite Lipodystrophy in Crebbp Heterozygous Mice. Nat. Genet. 2002, 30, 221–226. [Google Scholar] [CrossRef]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/Epigenetic Regulator CBP/P300 in Tumorigenesis: Structural and Functional Versatility in Target Recognition. Cell. Mol. Life Sci. 2013, 70, 3989–4008. [Google Scholar] [CrossRef]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of Histone Acetylation during Memory Formation in the Hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef] [Green Version]
- Monsey, M.S.; Ota, K.T.; Akingbade, I.F.; Hong, E.S.; Schafe, G.E. Epigenetic Alterations Are Critical for Fear Memory Consolidation and Synaptic Plasticity in the Lateral Amygdala. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Bousiges, O.; de Vasconcelos, A.P.; Neidl, R.; Cosquer, B.; Herbeaux, K.; Panteleeva, I.; Loeffler, J.-P.; Cassel, J.-C.; Boutillier, A.-L. Spatial Memory Consolidation Is Associated with Induction of Several Lysine-Acetyltransferase (Histone Acetyltransferase) Expression Levels and H2B/H4 Acetylation-Dependent Transcriptional Events in the Rat Hippocampus. Neuropsychopharmacology 2010, 35, 2521–2537. [Google Scholar] [CrossRef] [Green Version]
- Lubin, F.D.; Sweatt, J.D. The IκB Kinase Regulates Chromatin Structure during Reconsolidation of Conditioned Fear Memories. Neuron 2007, 55, 942–957. [Google Scholar] [CrossRef] [Green Version]
- Bredy, T.W.; Wu, H.; Crego, C.; Zellhoefer, J.; Sun, Y.E.; Barad, M. Histone Modifications around Individual BDNF Gene Promoters in Prefrontal Cortex are Associated with Extinction of Conditioned Fear. Learn. Mem. 2007, 14, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broide, R.S.; Redwine, J.M.; Aftahi, N.; Young, W.; Bloom, F.E.; Winrow, C.J. Distribution of Histone Deacetylases 1-11 in the Rat Brain. J. Mol. Neurosci. 2007, 31, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Duclot, F.; Meunier, J.; Naert, G.; Givalois, L.; Meffre, J.; Célérier, A.; Jacquet, C.; Copois, V.; Mechti, N.; et al. Altered Memory Capacities and Response to Stress in P300/CBP-Associated Factor (PCAF) Histone Acetylase Knockout Mice. Neuropsychopharmacology 2008, 33, 1584–1602. [Google Scholar] [CrossRef] [PubMed]
- Duclot, F.; Jacquet, C.; Gongora, C.; Maurice, T. Alteration of Working Memory but Not in Anxiety or Stress Response in P300/CBP Associated Factor (PCAF) Histone Acetylase Knockout Mice Bred on a C57BL/6 Background. Neurosci. Lett. 2010, 475, 179–183. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/P300 in Cell Growth, Transformation, and Development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [CrossRef]
- Yao, T.-P.; Oh, S.P.; Fuchs, M.; Zhou, N.-D.; Ch’ng, L.-E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene Dosage–Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator P300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Zou, X.; Watanabe, H.; van Deursen, J.M.; Shen, J. CBP Is Required for Both Short-Term and Long-Term Memory Formation. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 13066–13077. [Google Scholar] [CrossRef]
- Valor, L.M.; Pulopulos, M.M.; Jimenez-Minchan, M.; Olivares, R.; Lutz, B.; Barco, A. Ablation of CBP in Forebrain Principal Neurons Causes Modest Memory and Transcriptional Defects and a Dramatic Reduction of Histone Acetylation But Does Not Affect Cell Viability. J. Neurosci. 2011, 31, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Barco, A. The Rubinstein–Taybi Syndrome: Modeling Mental Impairment in the Mouse. Genes Brain Behav. 2007, 6, 32–39. [Google Scholar] [CrossRef]
- Korzus, E.; Rosenfeld, M.G.; Mayford, M. CBP Histone Acetyltransferase Activity Is a Critical Component of Memory Consolidation. Neuron 2004, 42, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.M.; Malvaez, M.; Kramar, E.; Matheos, D.P.; Arrizon, A.; Cabrera, S.M.; Lynch, G.; Greene, R.W.; Wood, M.A. Hippocampal Focal Knockout of CBP Affects Specific Histone Modifications, Long-Term Potentiation, and Long-Term Memory. Neuropsychopharmacology 2011, 36, 1545–1556. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.M.M.; Wood, M.A.; McDonough, C.B.; Abel, T. Transgenic Mice Expressing an Inhibitory Truncated Form of P300 Exhibit Long-Term Memory Deficits. Learn. Mem. 2007, 14, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, J.M.; Malleret, G.; Touzani, K.; Vronskaya, S.; Ishii, S.; Kandel, E.R.; Barco, A. Chromatin Acetylation, Memory, and LTP Are Impaired in CBP+/− Mice: A Model for the Cognitive Deficit in Rubinstein-Taybi Syndrome and Its Amelioration. Neuron 2004, 42, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Larizza, L.; Finelli, P. Developmental Disorders with Intellectual Disability Driven by Chromatin Dysregulation: Clinical Overlaps and Molecular Mechanisms. Clin. Genet. 2019, 95, 231–240. [Google Scholar] [CrossRef]
- Negri, G.; Magini, P.; Milani, D.; Crippa, M.; Biamino, E.; Piccione, M.; Sotgiu, S.; Perrìa, C.; Vitiello, G.; Frontali, M.; et al. Exploring by Whole Exome Sequencing Patients with Initial Diagnosis of Rubinstein–Taybi Syndrome: The Interconnections of Epigenetic Machinery Disorders. Hum. Genet. 2019, 138, 257–269. [Google Scholar] [CrossRef]
- Di Fede, E.; Massa, V.; Augello, B.; Squeo, G.; Scarano, E.; Perri, A.M.; Fischetto, R.; Causio, F.A.; Zampino, G.; Piccione, M.; et al. Expanding the Phenotype Associated to KMT2A Variants: Overlapping Clinical Signs between Wiedemann–Steiner and Rubinstein–Taybi Syndromes. Eur. J. Hum. Genet. 2020, 1–11. [Google Scholar] [CrossRef]
- Woods, S.A.; Robinson, H.B.; Kohler, L.J.; Agamanolis, D.; Sterbenz, G.; Khalifa, M. Exome Sequencing Identifies a Novel EP300 Frame Shift Mutation in a Patient with Features That Overlap Cornelia de Lange Syndrome. Am. J. Med. Genet. A 2014, 164, 251–258. [Google Scholar] [CrossRef]
- Cucco, F.; Sarogni, P.; Rossato, S.; Alpa, M.; Patimo, A.; Latorre, A.; Magnani, C.; Puisac, B.; Ramos, F.J.; Pié, J.; et al. Pathogenic Variants in EP300 and ANKRD11 in Patients with Phenotypes Overlapping Cornelia de Lange Syndrome. Am. J. Med. Genet. A 2020, 182, 1690–1696. [Google Scholar] [CrossRef]
- Sarogni, P.; Pallotta, M.M.; Musio, A. Cornelia de Lange Syndrome: From Molecular Diagnosis to Therapeutic Approach. J. Med. Genet. 2020, 57, 289–295. [Google Scholar] [CrossRef]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and Management of Cornelia de Lange Syndrome: First International Consensus Statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Tipping the Balance of Chromatin States. Annu. Rev. Genomics Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.-C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Bjornsson, H.T. The Mendelian Disorders of the Epigenetic Machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer Activities of Histone Deacetylase Inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Gräff, J.; Tsai, L.-H. The Potential of HDAC Inhibitors as Cognitive Enhancers. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 311–330. [Google Scholar] [CrossRef] [Green Version]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T.; et al. Histone Deacetylase Inhibitors Enhance Memory and Synaptic Plasticity via CREB:CBP-Dependent Transcriptional Activation. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [CrossRef]
- Wood, M.A.; Kaplan, M.P.; Park, A.; Blanchard, E.J.; Oliveira, A.M.M.; Lombardi, T.L.; Abel, T. Transgenic Mice Expressing a Truncated Form of CREB-Binding Protein (CBP) Exhibit Deficits in Hippocampal Synaptic Plasticity and Memory Storage. Learn. Mem. 2005, 12, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haettig, J.; Stefanko, D.P.; Multani, M.L.; Figueroa, D.X.; McQuown, S.C.; Wood, M.A. HDAC Inhibition Modulates Hippocampus-Dependent Long-Term Memory for Object Location in a CBP-Dependent Manner. Learn. Mem. 2011, 18, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Babu, A.; Kamaraj, M.; Basu, M.; Mukherjee, D.; Kapoor, S.; Ranjan, S.; Swamy, M.M.; Kaypee, S.; Scaria, V.; Kundu, T.K.; et al. Chemical and Genetic Rescue of an Ep300 Knockdown Model for Rubinstein Taybi Syndrome in Zebrafish. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 1203–1215. [Google Scholar] [CrossRef]
- Lopez-Atalaya, J.P.; Gervasini, C.; Mottadelli, F.; Spena, S.; Piccione, M.; Scarano, G.; Selicorni, A.; Barco, A.; Larizza, L. Histone Acetylation Deficits in Lymphoblastoid Cell Lines from Patients with Rubinstein–Taybi Syndrome. J. Med. Genet. 2012, 49, 66–74. [Google Scholar] [CrossRef]
- Parodi, C.; Di Fede, E.; Peron, A.; Viganò, I.; Grazioli, P.; Castiglioni, S.; Finnell, R.H.; Gervasini, C.; Vignoli, A.; Massa, V. Chromatin Imbalance as the Vertex Between Fetal Valproate Syndrome and Chromatinopathies. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Schölz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/P300 Acetylome. Cell 2018, 174, 231.e12–244.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotypic Features | CREBBP (n = 422) | EP300 (n = 74) | ||
|---|---|---|---|---|
| Percentage | Number | Percentage | Number | |
| Intrauterine growth retardation | 25 | 55/220 | 43.1 | 25/58 |
| Preeclampsia | 3.4 | 2/59 | 25 | 16/64 |
| Postnatal growth retardation | 62.3 | 203/326 | 59.7 | 43/72 |
| Microcephaly | 52.7 | 129/245 | 82.4 | 61/74 |
| Hypertrichosis | 76.4 | 123/161 | 47.4 | 27/57 |
| Facial dysmorphism | ||||
| Arched eyebrows | 85.6 | 119/139 | 65.6 | 42/64 |
| Long eyelashes | 88.6 | 109/123 | 83.6 | 51/61 |
| Downslanted palpebral fissures | 81.1 | 258/318 | 51.6 | 33/64 |
| Beaked nose | 81.7 | 272/333 | 37.5 | 24/64 |
| Columella below alae nasi | 87.4 | 228/261 | 82.8 | 53/64 |
| Highly arched palate | 79.8 | 197/247 | 56.1 | 32/57 |
| Micrognathia | 64.2 | 149/232 | 40.6 | 26/64 |
| Grimacing smile | 94.9 | 112/118 | 36.8 | 21/57 |
| Low-set ears | 51.1 | 112/219 | 23.4 | 15/64 |
| Broad thumbs/halluces | 92.3 | 373/404 | 59.5 | 44/74 |
| Angulated thumbs | 56.4 | 184/326 | 4.8 | 3/63 |
| Intellectual disability | 82.2 | 287/349 | 84.9 | 62/73 |
| Severe | 35.9 | 33/92 | 7.3 | 3/41 |
| Moderate | 47.8 | 44/92 | 26.8 | 11/41 |
| Mild | 14.1 | 13/92 | 65.9 | 27/41 |
| Autism/Behavioral problems | 49.4 | 78/158 | 21.3 | 13/61 |
| Cardiovascular anomalies | 34.5 | 99/287 | 29 | 20/69 |
| Urinary tract anomalies | 37.4 | 61/163 | 26.3 | 15/57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Gils, J.; Magdinier, F.; Fergelot, P.; Lacombe, D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes 2021, 12, 968. https://doi.org/10.3390/genes12070968
Van Gils J, Magdinier F, Fergelot P, Lacombe D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes. 2021; 12(7):968. https://doi.org/10.3390/genes12070968
Chicago/Turabian StyleVan Gils, Julien, Frederique Magdinier, Patricia Fergelot, and Didier Lacombe. 2021. "Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder" Genes 12, no. 7: 968. https://doi.org/10.3390/genes12070968
APA StyleVan Gils, J., Magdinier, F., Fergelot, P., & Lacombe, D. (2021). Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes, 12(7), 968. https://doi.org/10.3390/genes12070968