



Genes 2025, 16(1), 104; https://doi.org/10.3390/genes16010104 - 19 Jan 2025
Viewed by 853
Abstract
►
Show Figures
Mendelian disorders of the epigenetic machinery (MDEMs) include a large number of conditions caused by defective activity of a member of the epigenetic machinery. MDEMs are characterized by multiple congenital abnormalities, intellectual disability and abnormal growth. that can be variably up- or down-regulated.
[...] Read more.
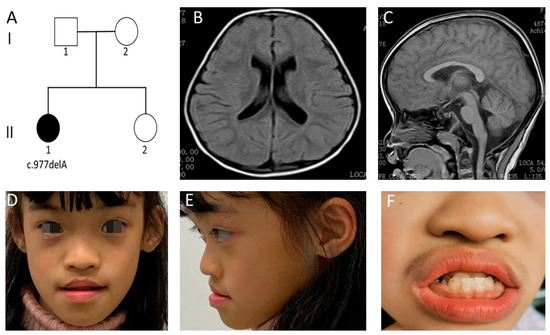
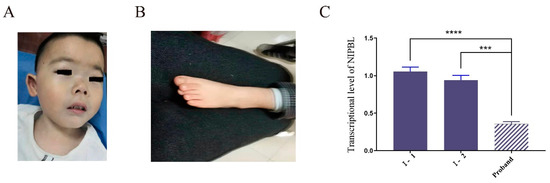
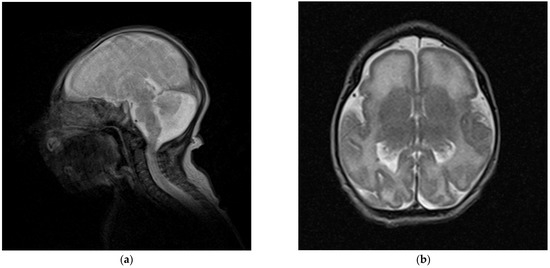
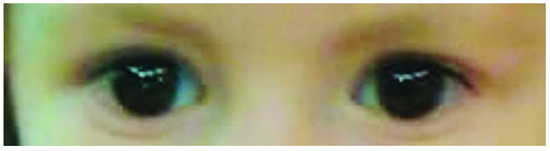
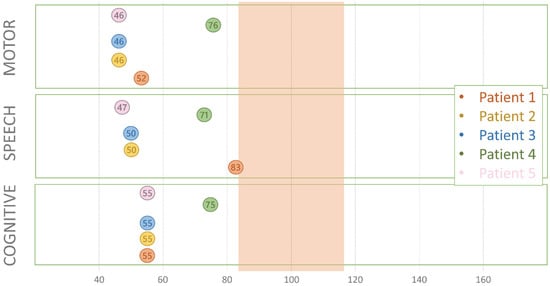
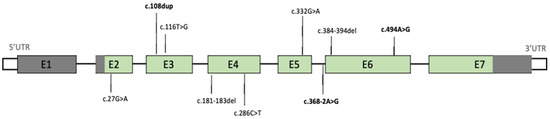
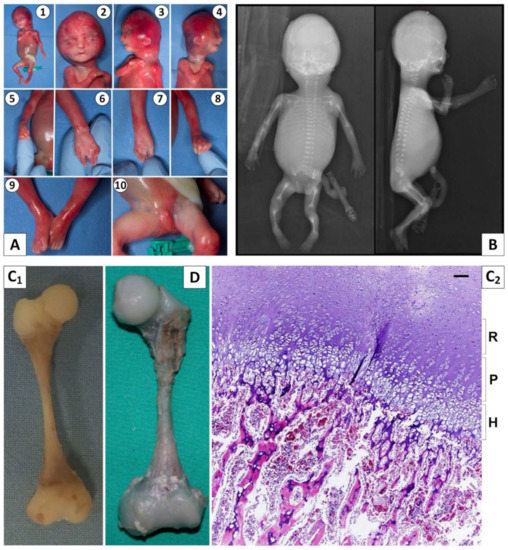
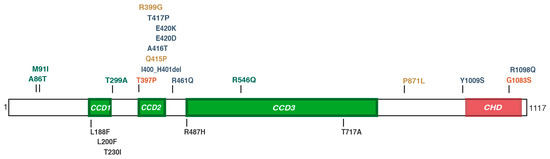
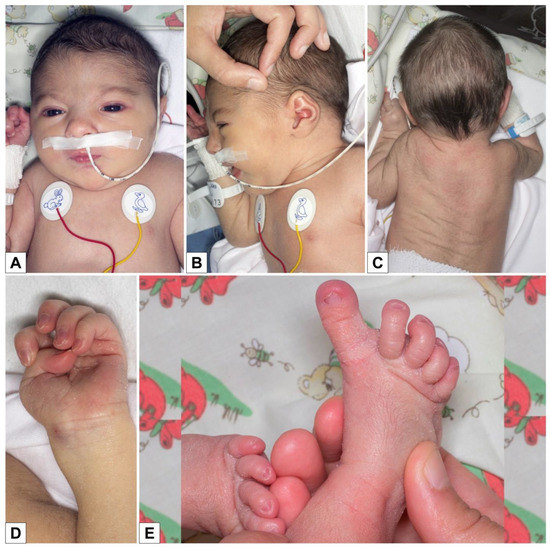
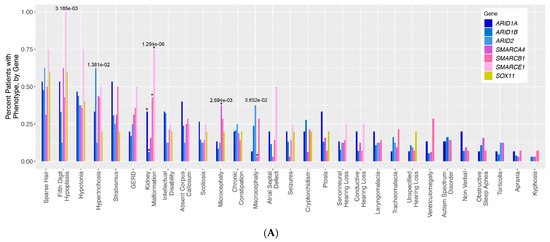
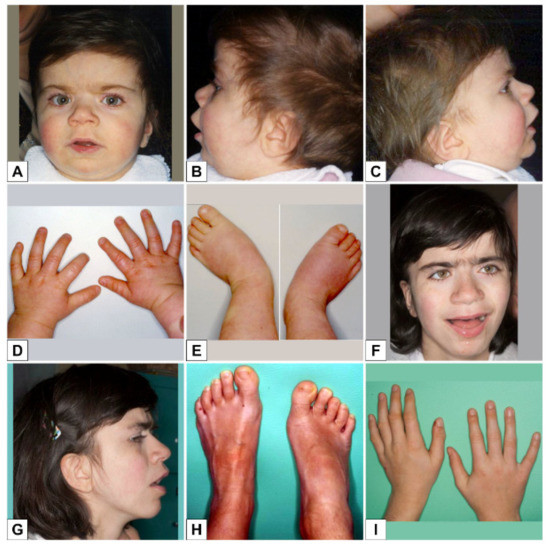
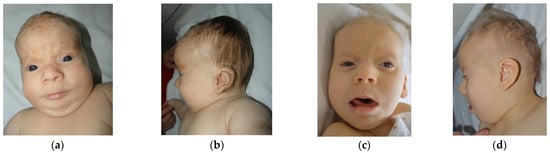
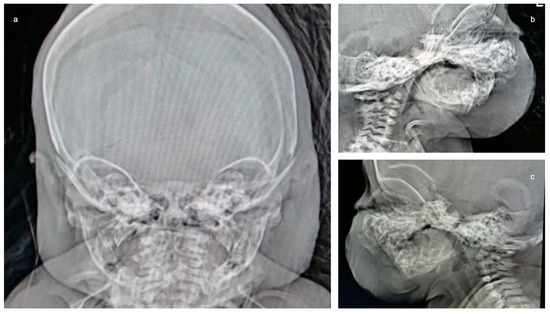
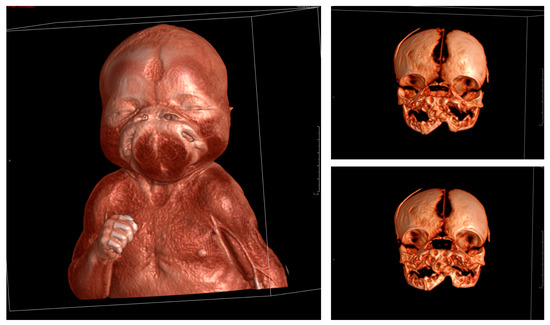
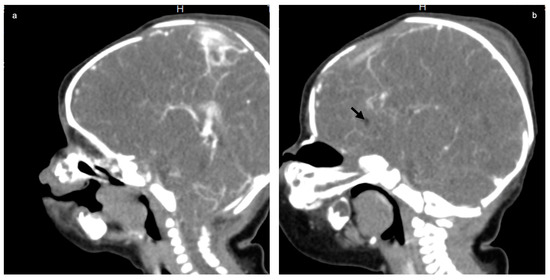
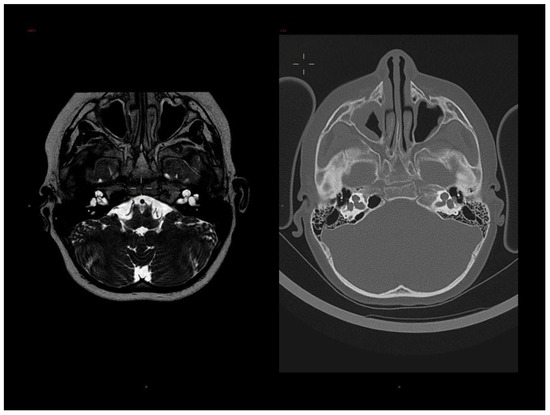
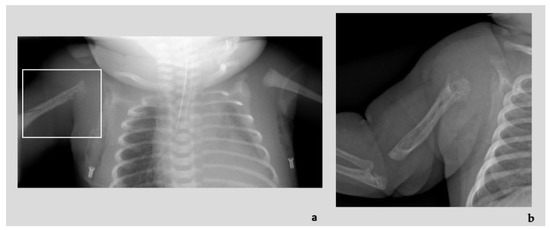
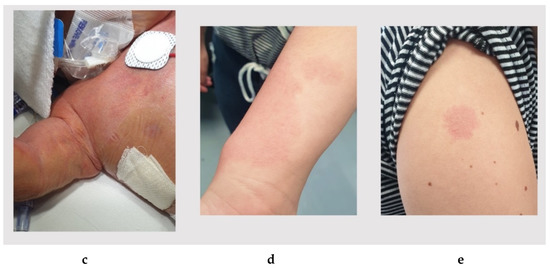
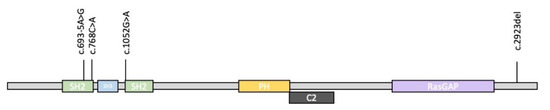
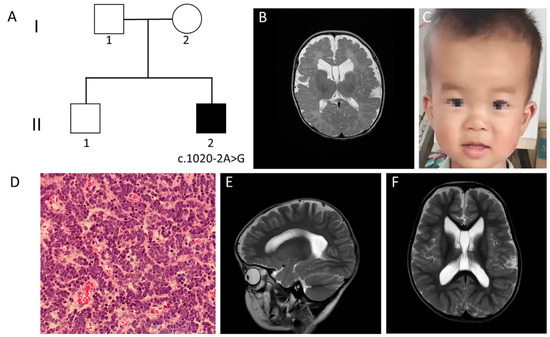
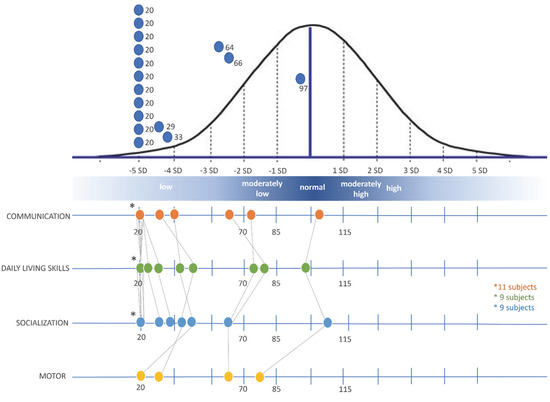
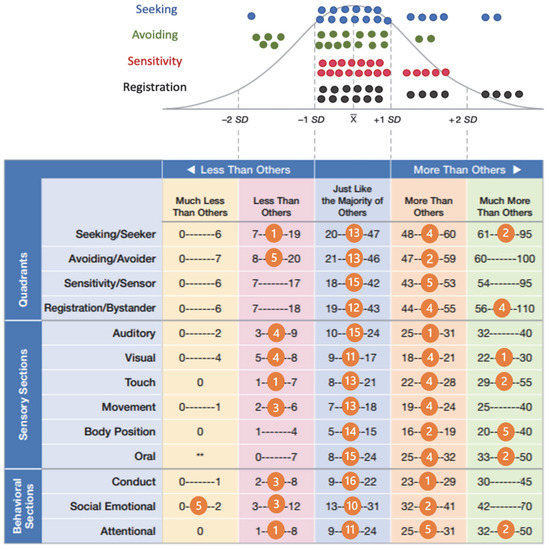
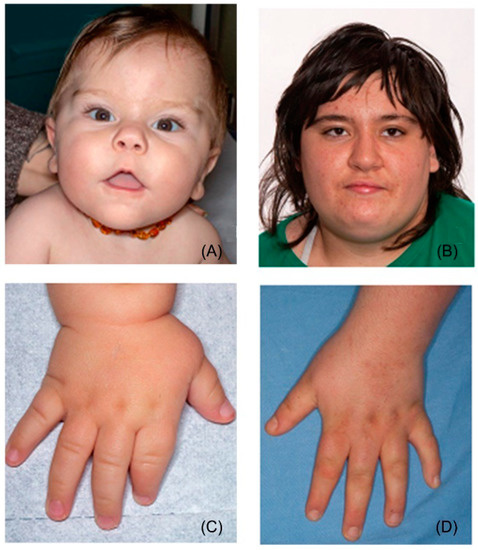
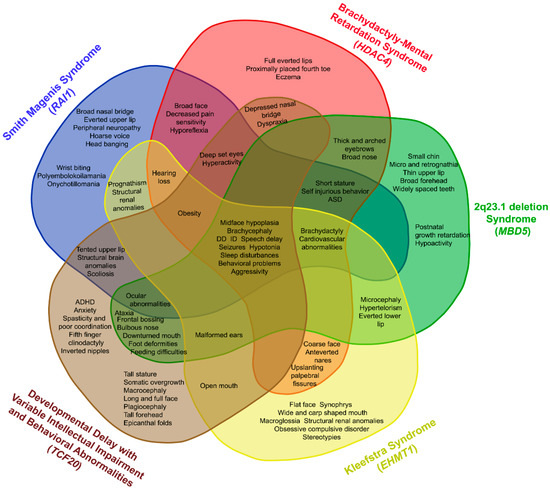
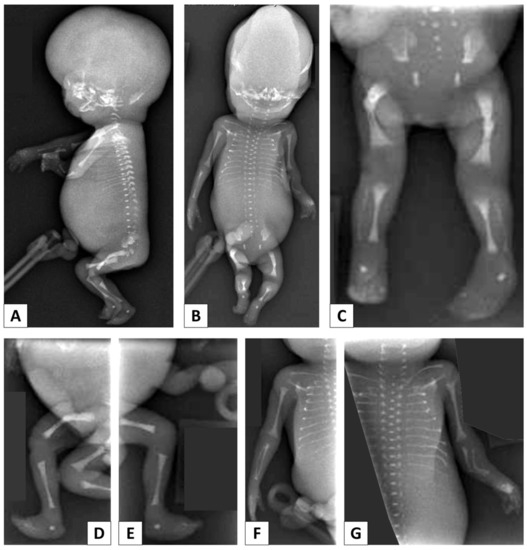
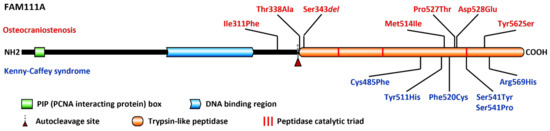
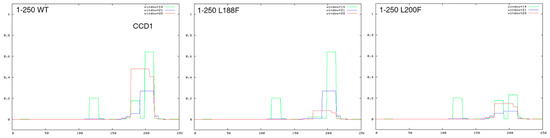
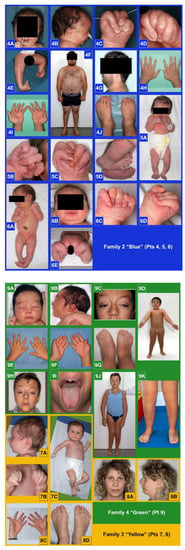
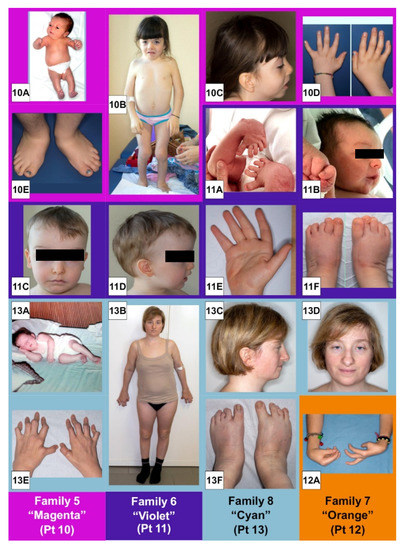
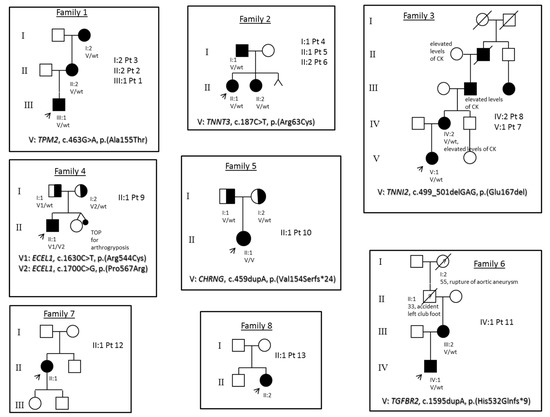
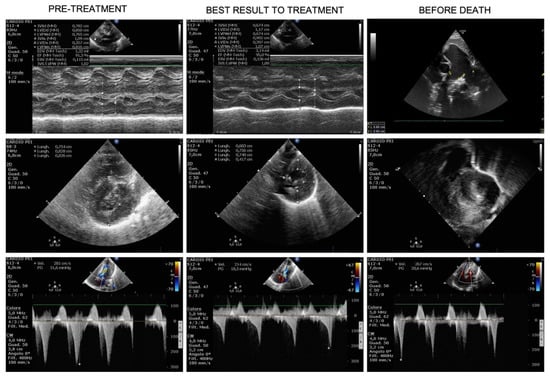
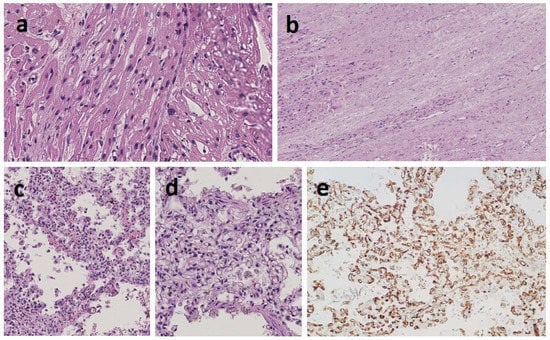
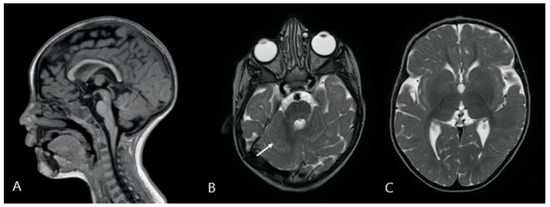
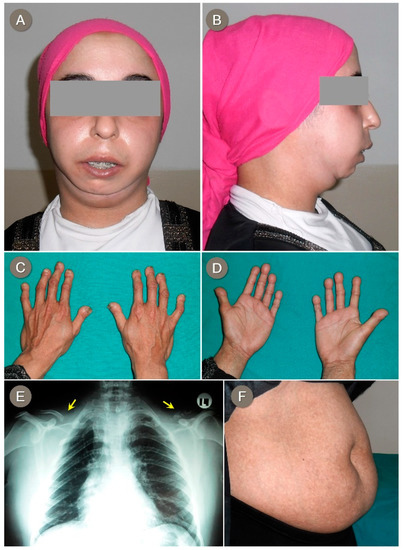
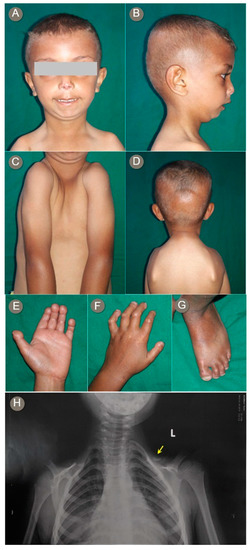
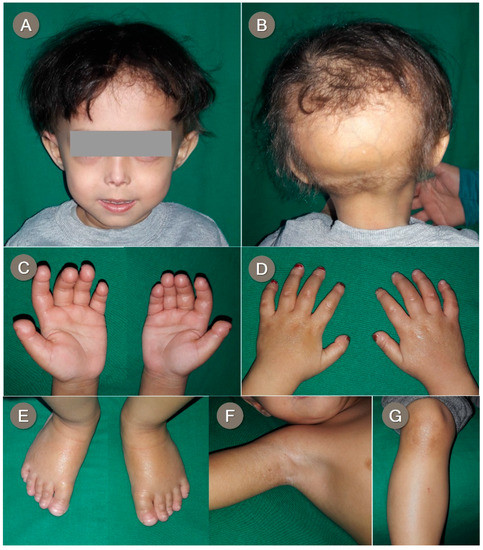
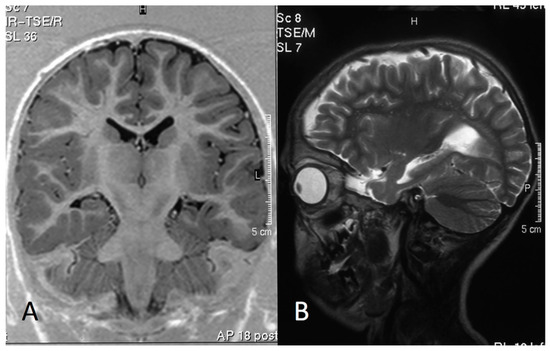
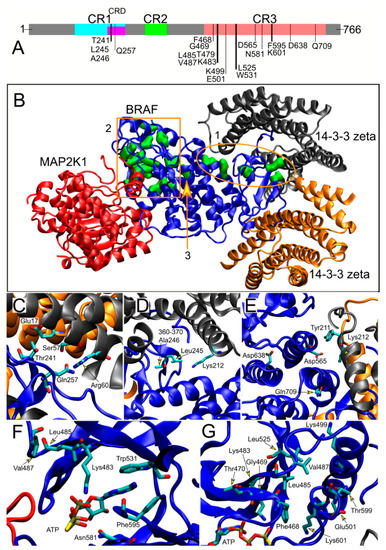
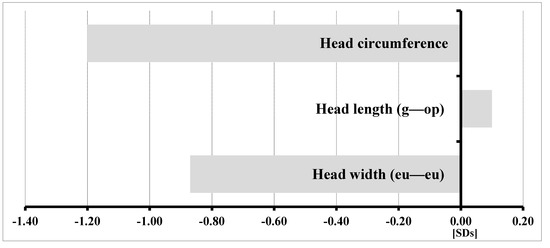
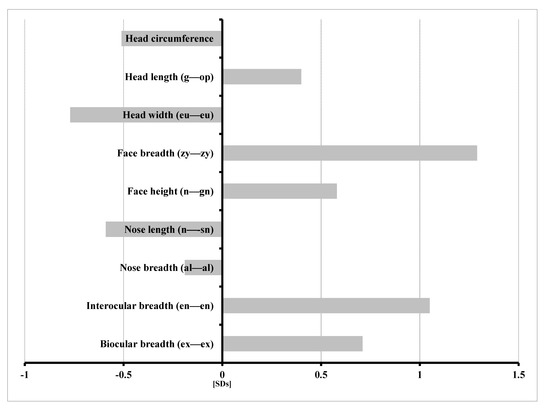
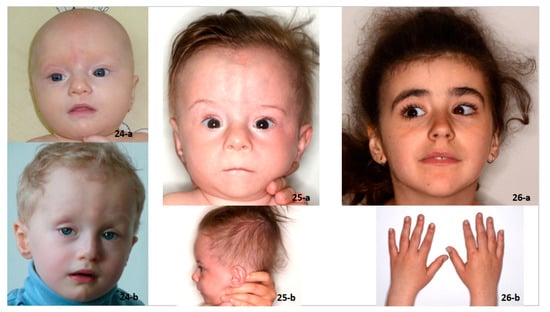
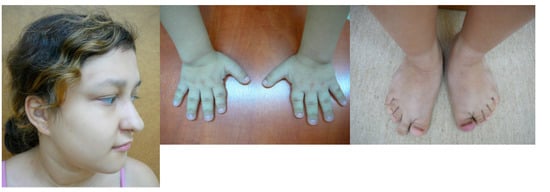
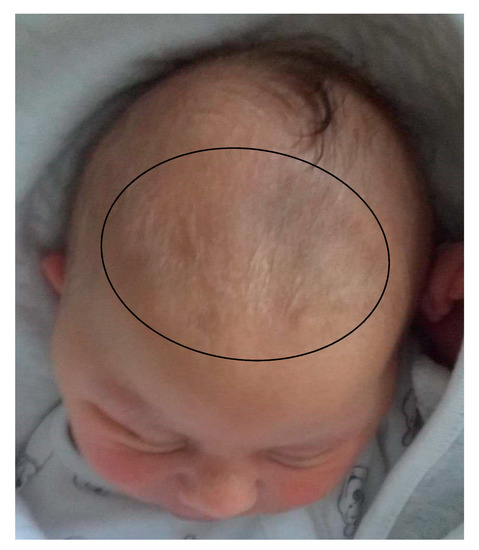
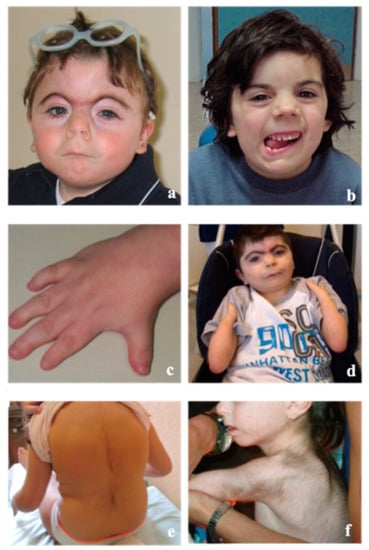
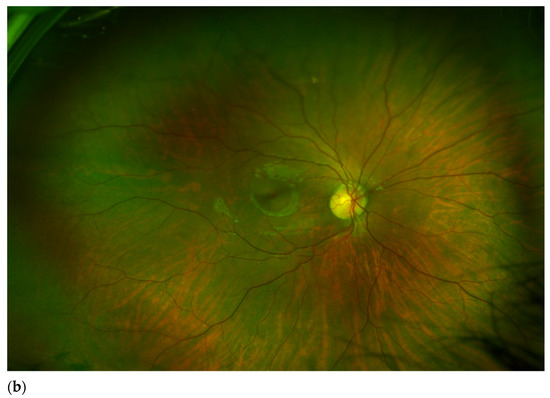
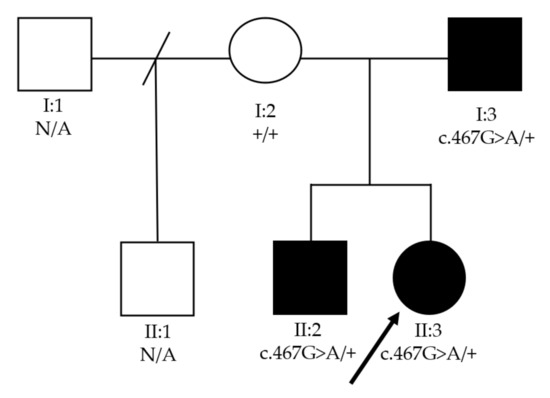
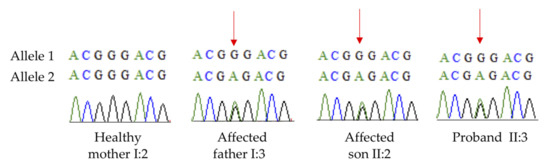
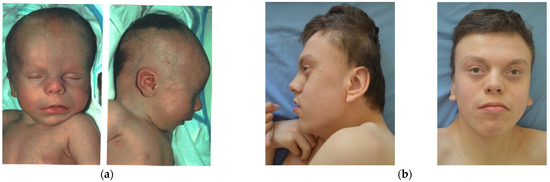
Mendelian disorders of the epigenetic machinery (MDEMs) include a large number of conditions caused by defective activity of a member of the epigenetic machinery. MDEMs are characterized by multiple congenital abnormalities, intellectual disability and abnormal growth. that can be variably up- or down-regulated. Background/Objectives: In several MDEMs, a predisposition to metabolic syndrome and obesity since childhood has been reported. Methods: To investigate the metabolic bases of this abnormal growth, we collected physical data from a heterogeneous pool of 38 patients affected by MDEMs. Thirty-five patients performed indirect calorimetry (as a measure of resting energy expenditure, REE) and blood tests to monitor plasmatic nutritional parameters. Conclusions: Although limited by a small-sized and heterogeneous sample, our study demonstrates a linear correlation between REE and physical parameters, OFC, height and weight, and observed a slight imbalance on several plasmatic spies of metabolic syndrome predisposition. Furthermore, we demonstrated a significantly higher REE in Sotos Syndrome type 1 patients compared to the controls, which resulted independent from height, suggesting that impaired metabolism in these patients may go beyond overgrowth.
Full article

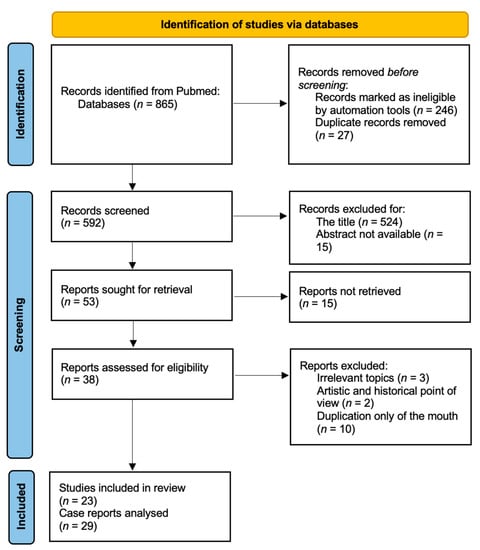
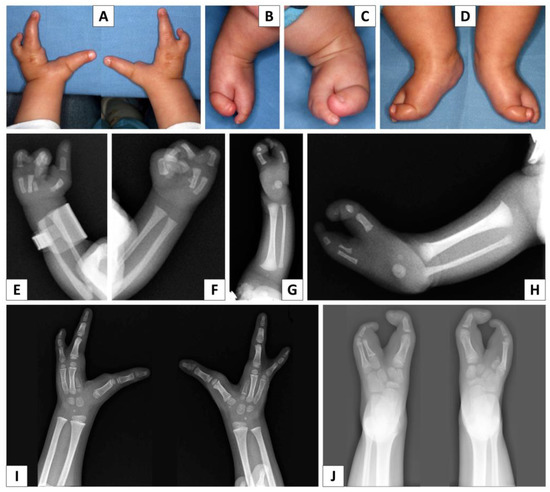
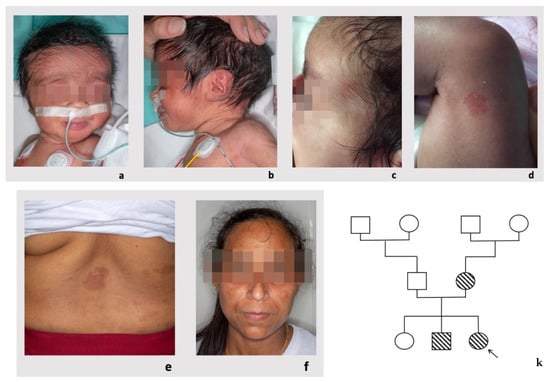
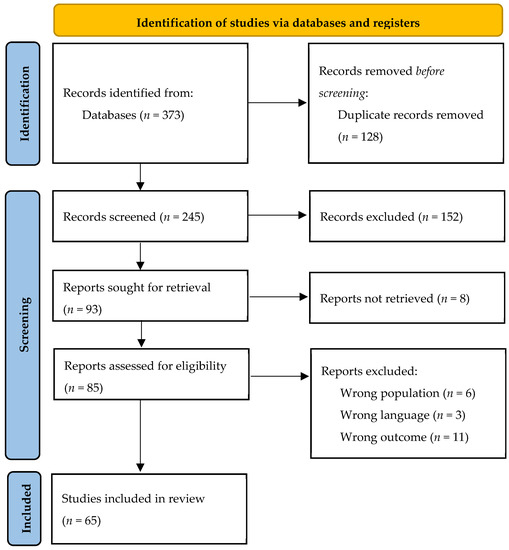
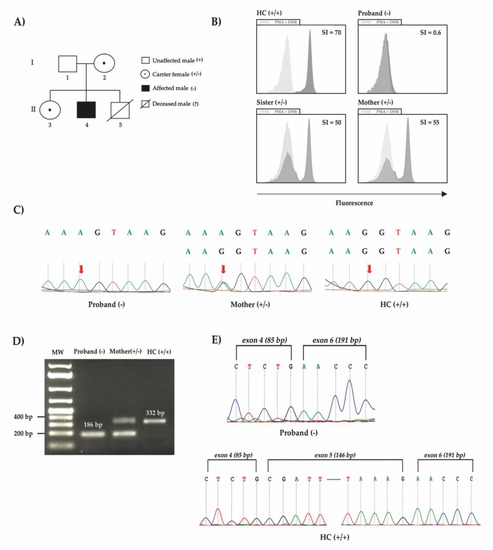
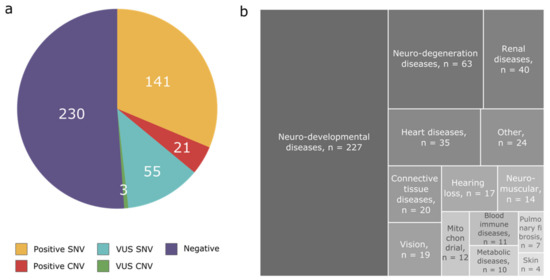
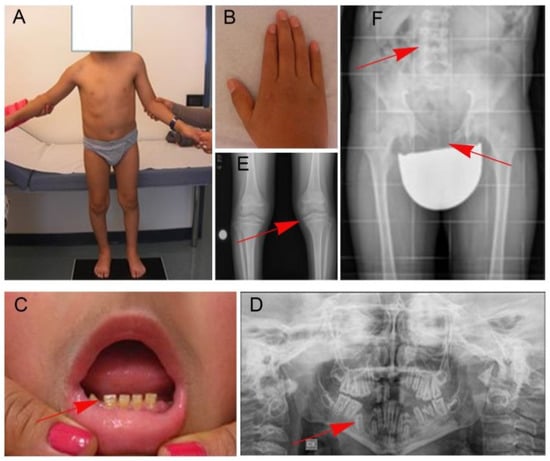
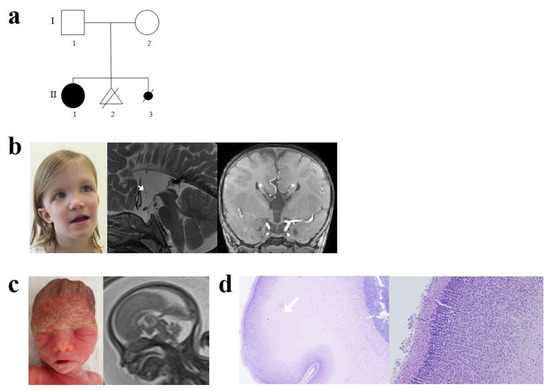
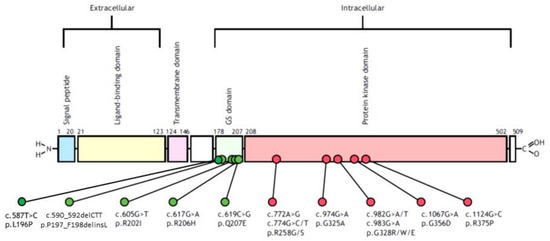
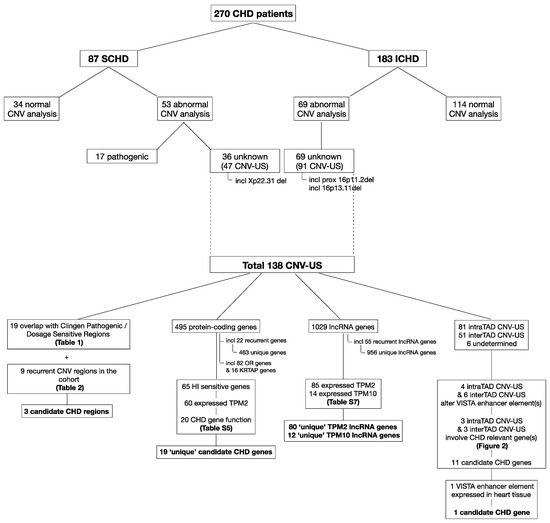
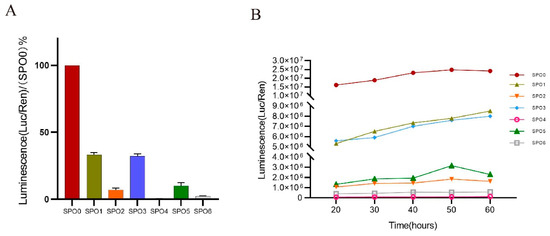
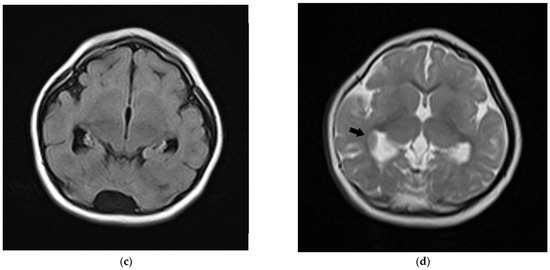
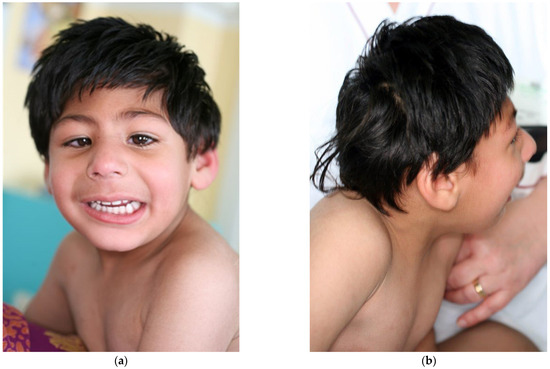
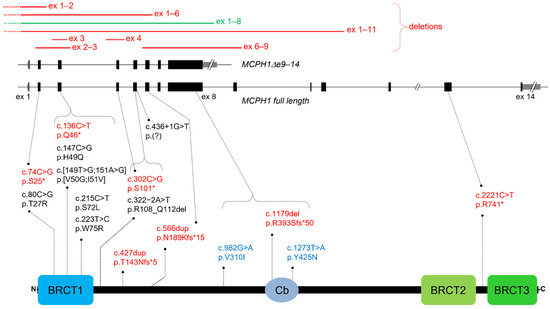
Figure 1





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}