
Set1 Targets Genes with Essential Identity and Tumor-Suppressing Functions in Planarian Stem Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mnase ChIP-seq
2.2. Analysis of ChIP-seq Datasets
2.3. Planarian Culture, RNAi and Radiation
2.4. Whole-Mount In Situ Hybridization and Immunofluorescence
3. Results
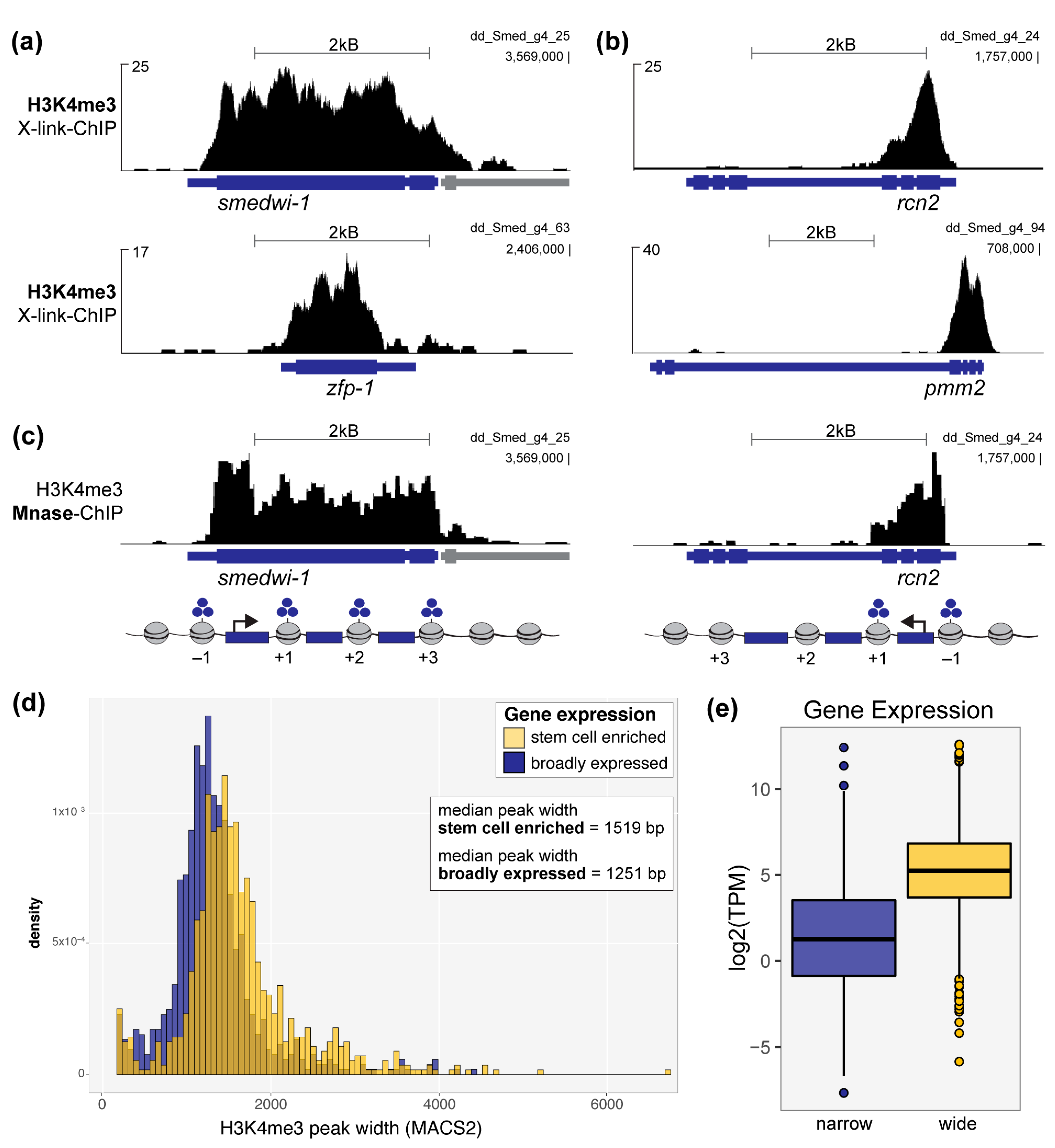
3.1. A Broad Histone H3K4me3 Chromatin Signature Is Conserved in Planarians
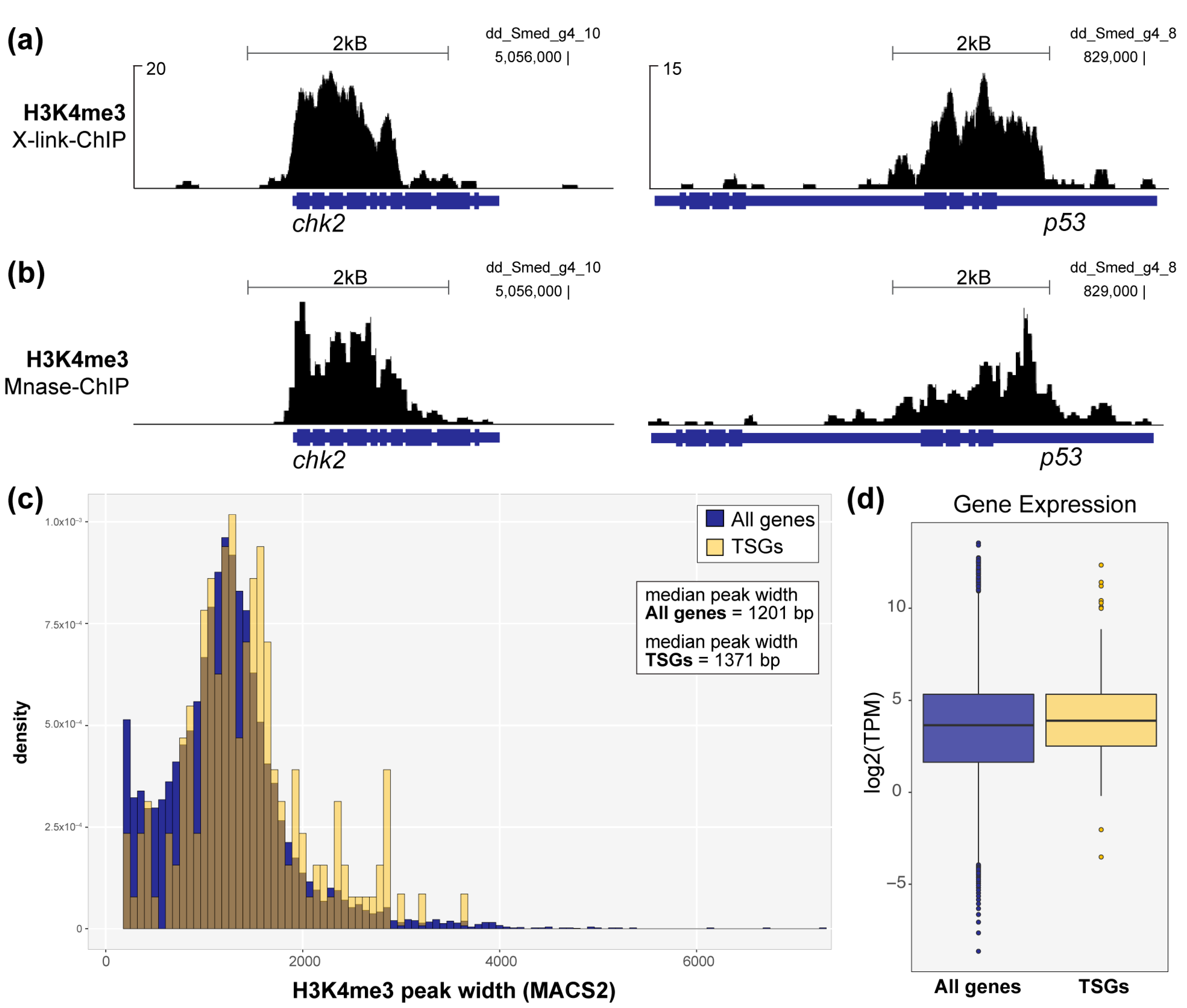
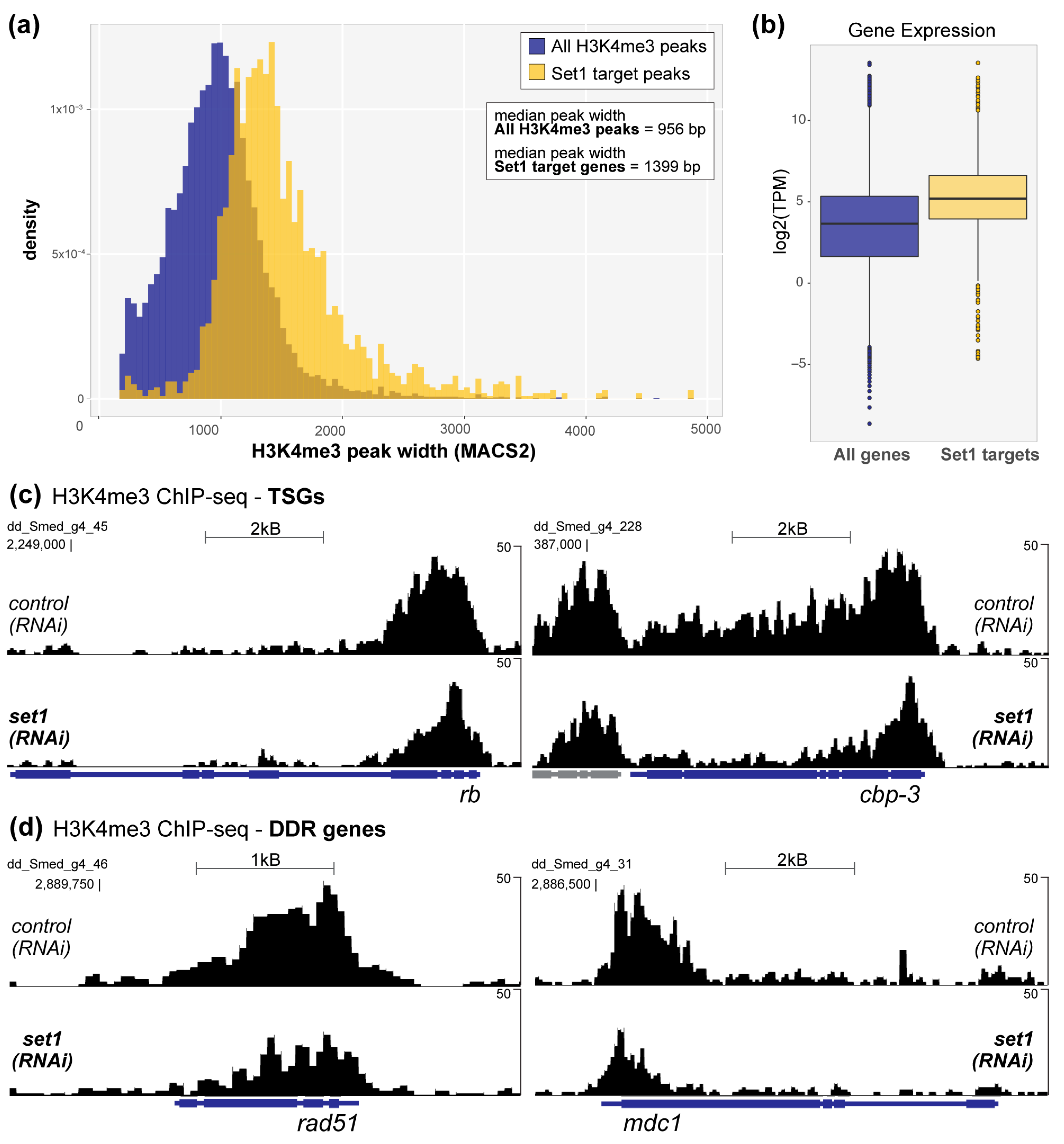
3.2. Set1 Targets TSGs in Planarian Stem Cells
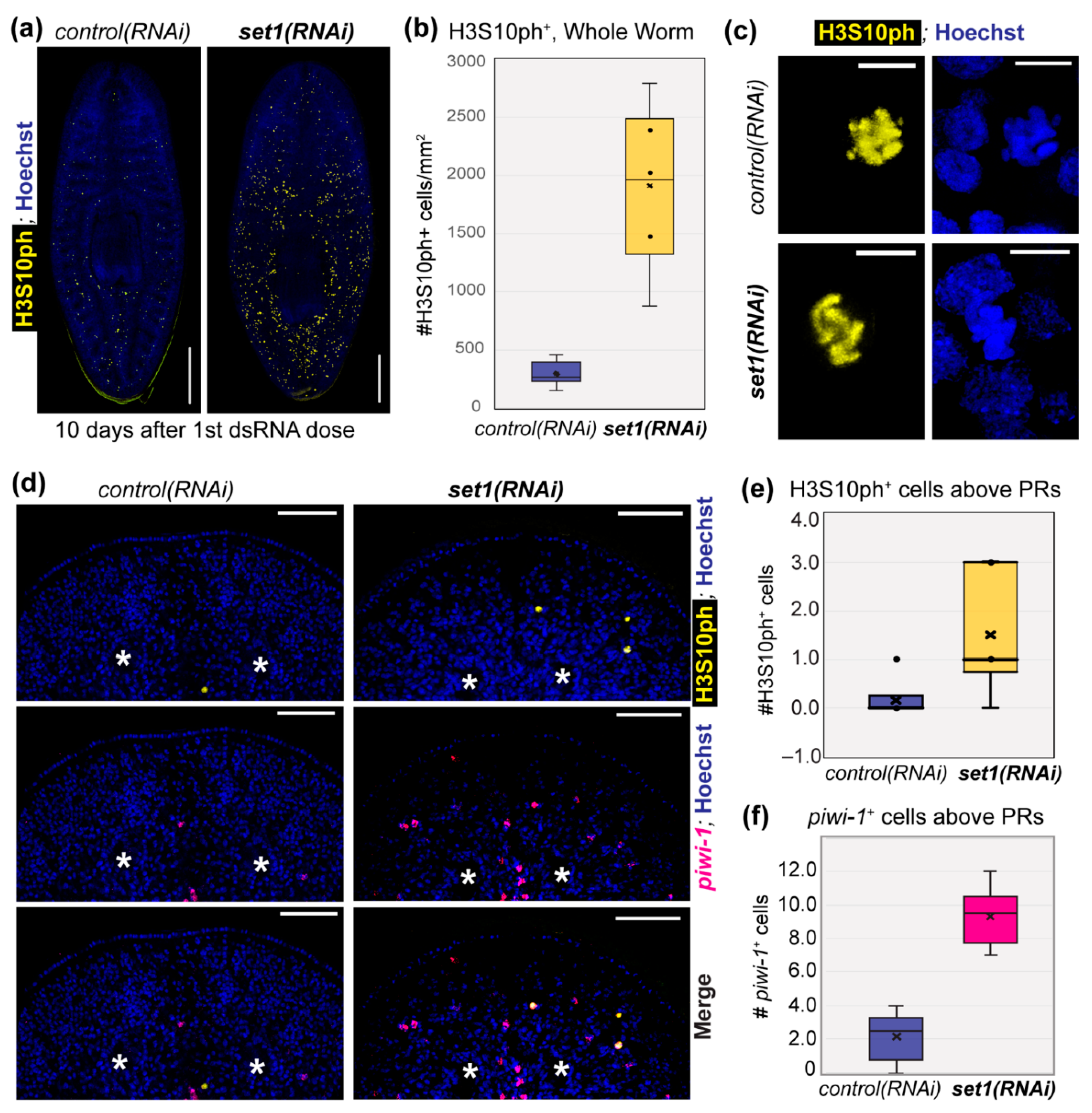
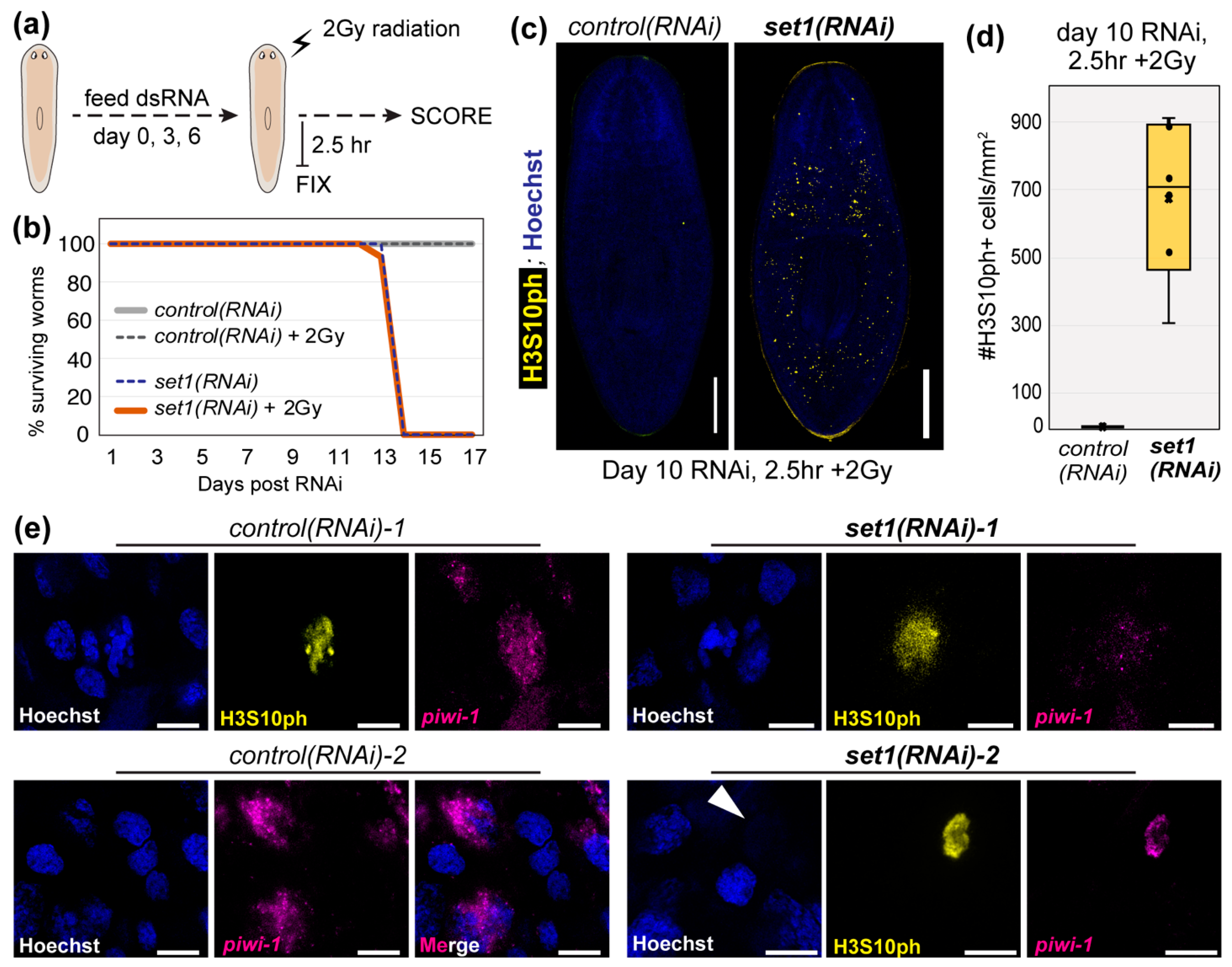
3.3. Set1 Is Required for Tumor-Suppressing Functions in Planarians
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lei, K.; Vu, H.; Mohan, R.D.; McKinney, S.A.; Seidel, C.W.; Alexander, R.; Gotting, K.; Workman, J.L.; Alvarado, A.S. Egf Signaling Directs Neoblast Repopulation by Regulating Asymmetric Cell Division in Planarians. Dev. Cell 2016, 38, 413–429. [Google Scholar] [CrossRef] [Green Version]
- Oviedo, N.J.; Pearson, B.; Levin, M.; Alvarado, A.S. Planarian PTEN homologs regulate stem cells and regeneration through TOR signaling. Dis. Model. Mech. 2008, 1, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Pearson, B.; Alvarado, A.S. A planarian p53 homolog regulates proliferation and self-renewal in adult stem cell lineages. Development 2010, 137, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, Y.; Abnave, P.; Kao, D.; Hughes, S.; Lai, A.; Jaber-Hijazi, F.; Kosaka, N.; Aboobaker, A.A. Conservation of epigenetic regulation by the MLL3/4 tumour suppressor in planarian pluripotent stem cells. Nat. Commun. 2018, 9, 3633. [Google Scholar] [CrossRef] [Green Version]
- Lindsay-Mosher, N.; Chan, A.; Pearson, B.J. Planarian EGF repeat-containing genes megf6 and hemicentin are required to restrict the stem cell compartment. PLoS Genet. 2020, 16, e1008613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, E.M.; Chitsazan, A.D.; Seidel, C.W.; Alvarado, A.S. Set1 and MLL1/2 Target Distinct Sets of Functionally Different Genomic Loci in Vivo. Cell Rep. 2015, 13, 2741–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, A.; Henderson, J.M.; Ross, K.G.; Cowles, M.W.; Torres, J.; Zayas, R.M. Epigenetic regulation of planarian stem cells by the SET1/MLL family of histone methyltransferases. Epigenetics 2013, 8, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Önal, P.; Grün, D.; Adamidi, C.; Rybak-Wolf, A.; Solana, J.; Mastrobuoni, G.; Wang, Y.; Rahn, H.-P.; Chen, W.; Kempa, S.; et al. Gene expression of pluripotency determinants is conserved between mammalian and planarian stem cells. EMBO J. 2012, 31, 2755–2769. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.E.; Ho, J.; Reddien, P.W. Genetic regulators of a pluripotent adult stem cell system in planarians identified by rnai and clonal analysis. Cell Stem Cell 2012, 10, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Chen, Z.; Wu, D.; Zhang, L.; Lin, X.; Su, J.; Rodriguez, B.; Xi, Y.; Xia, Z.; Chen, X.; et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet. 2015, 47, 1149–1157. [Google Scholar] [CrossRef]
- Hayashi, T.; Asami, M.; Higuchi, S.; Shibata, N.; Agata, K. Isolation of planarian X-ray-sensitive stem cells by fluorescence-activated cell sorting. Dev. Growth Differ. 2006, 48, 371–380. [Google Scholar] [CrossRef]
- Romero, B.T.; Evans, D.J.; Aboobaker, A.A. FACS analysis of the planarian stem cell compartment as a tool to understand regenerative mechanisms. In Progenitor Cells; Humana Press: Totowa, NJ, USA, 2012; pp. 167–179. [Google Scholar] [CrossRef]
- Brand, M.; Rampalli, S.; Chaturvedi, C.-P.; Dilworth, F.J. Analysis of epigenetic modifications of chromatin at specific gene loci by native chromatin immunoprecipitation of nucleosomes isolated using hydroxyapatite chromatography. Nat. Protoc. 2008, 3, 398–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurmann, W.; Peter, R. Planarian cell culture: A comparative review of methods and an improved protocol for primary cultures of neoblasts. Belg. J. Zool. 2001, 131, 123–130. [Google Scholar]
- Grohme, M.; Schloissnig, S.; Rozanski, A.; Pippel, M.; Young, G.; Winkler, S.; Brandl, H.; Henry, I.; Dahl, A.; Powell, S.; et al. The genome of Schmidtea mediterranea and the evolution of core cellular mechanisms. Nature 2018, 554, 56–61. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Shao, N.Y.; Liu, X.; Maze, I.; Feng, J.; Nestler, E.J. diffReps: Detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS ONE 2013, 8, e65598. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Merryman, M.S.; Alvarado, A.S.; Jenkin, J.C. Culturing Planarians in the Laboratory. Methods Mol. Biol. 2018, 1774, 241–258. [Google Scholar] [CrossRef]
- Rouhana, L.; Weiss, J.A.; Forsthoefel, D.; Lee, H.; King, R.S.; Inoue, T.; Shibata, N.; Agata, K.; Newmark, P. RNA interference by feeding in vitro-synthesized double-stranded RNA to planarians: Methodology and dynamics. Dev. Dyn. 2013, 242, 718–730. [Google Scholar] [CrossRef] [Green Version]
- Forsthoefel, D.J.; Ross, K.G.; Newmark, P.A.; Zayas, R.M. Fixation, Processing, and Immunofluorescent Labeling of Whole Mount Planarians. Methods Mol. Biol. 2018, 1774, 353–366. [Google Scholar]
- King, R.S.; Newmark, P.A. Whole-Mount in Situ Hybridization of Planarians. Methods Mol. Biol. 2018, 1774, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Shiroor, D.A.; Bohr, T.E.; Adler, C.E. Injury Delays Stem Cell Apoptosis after Radiation in Planarians. Curr. Biol. 2020, 30, 2166–2174.e3. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.; Kulbokas, E.J.; Gingeras, T.; et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Benayoun, B.A.; Pollina, E.A.; Ucar, D.; Mahmoudi, S.; Karra, K.; Wong, E.D.; Devarajan, K.; Daugherty, A.C.; Kundaje, A.; Mancini, E.; et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 2014, 158, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Benayoun, B.A.; Pollina, E.A.; Ucar, D.; Mahmoudi, S.; Karra, K.; Wong, E.D.; Devarajan, K.; Daugherty, A.C.; Kundaje, A.B.; Mancini, E.; et al. H3K4me3 Breadth Is Linked to Cell Identity and Transcriptional Consistency. Cell 2015, 163, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of Histone H3K4 Methylation Patterns. Mol. Cell 2017, 68, 773–785.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resch, A.M.; Palakodeti, D.; Lu, Y.-C.; Horowitz, M.; Graveley, B.R. Transcriptome Analysis Reveals Strain-Specific and Conserved Stemness Genes in Schmidtea mediterranea. PLoS ONE 2012, 7, e34447. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Klein, D.C.; Hainer, J. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 2020, 28, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Rozanski, A.; Moon, H.; Brandl, H.; Martin-Duran, J.M.; Grohme, M.A.; Huttner, K.; Bartscherer, K.; Henry, I.; Rink, J.C. PlanMine 3.0-improvements to a mineable resource of flatworm biology and biodiversity. Nucleic Acids Res. 2019, 47, D812–D820. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Hariharan, I.K.; Bilder, D. Regulation of imaginal disc growth by tumor-suppressor genes in Drosophila. Annu. Rev. Genet. 2006, 40, 335–361. [Google Scholar] [CrossRef]
- Jafarnejad, S.M.; Li, G. Regulation of p53 by ING family members in suppression of tumor initiation and progression. Cancer Metastasis Rev. 2012, 31, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.T.; Tajmir, P.; Lin, C.H.; Liadis, N.; Zhu, X.D.; Eweida, M.; Tolasa-Karaman, G.; Cai, F.; Wang, R.; Kitamura, T.; et al. Essential role of Pten in body size determination and pancreatic beta-cell homeostasis in vivo. Mol. Cell. Biol. 2006, 26, 4511–4518. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, Y.; Sato, R.; Akiyama, T. Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat. Cell Biol. 2003, 5, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Marian, T.A.; Lee, H.; Kodama, M.; Li, J.; Parmacek, M.S.; Jenkins, N.A.; Copeland, N.G.; Wei, Z. MRTFB suppresses colorectal cancer development through regulating SPDL1 and MCAM. Proc. Natl. Acad. Sci. USA 2019, 116, 23625–23635. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Wagner, D.E.; Wang, I.E.; Reddien, P.W. Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 2011, 332, 811–816. [Google Scholar] [CrossRef]
- Sharma, A.K.; Bhattacharya, S.; Khan, S.A.; Khade, B.; Gupta, S. Dynamic alteration in H3 serine 10 phosphorylation is G1-phase specific during ionization radiation induced DNA damage response in human cells. Mutat. Res. 2015, 773, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Niida, H.; Zineldeen, D.; Tagami, H.; Tanaka, M.; Saito, H.; Nakanishi, M. Chk1 is a histone h3 threonine 11 kinase that regulates dna damage-induced transcriptional repression. Cell 2008, 132, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R.; de La Pompa, J.L.; Sirard, C.; Mo, R.; Woo, M.; Hakem, A.; Wakeham, A.; Potter, J.; Reitmair, A.; Billia, F.; et al. The tumor suppressor gene brca1 is required for embryonic cellular proliferation in the mouse. Cell 1996, 85, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, C.; Xu, X.; Deng, C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3667–3672. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, P.; Waterbury, C.K.M.; Duncan, E.M. Set1 Targets Genes with Essential Identity and Tumor-Suppressing Functions in Planarian Stem Cells. Genes 2021, 12, 1182. https://doi.org/10.3390/genes12081182
Verma P, Waterbury CKM, Duncan EM. Set1 Targets Genes with Essential Identity and Tumor-Suppressing Functions in Planarian Stem Cells. Genes. 2021; 12(8):1182. https://doi.org/10.3390/genes12081182
Chicago/Turabian StyleVerma, Prince, Court K. M. Waterbury, and Elizabeth M. Duncan. 2021. "Set1 Targets Genes with Essential Identity and Tumor-Suppressing Functions in Planarian Stem Cells" Genes 12, no. 8: 1182. https://doi.org/10.3390/genes12081182
APA StyleVerma, P., Waterbury, C. K. M., & Duncan, E. M. (2021). Set1 Targets Genes with Essential Identity and Tumor-Suppressing Functions in Planarian Stem Cells. Genes, 12(8), 1182. https://doi.org/10.3390/genes12081182