
Whole-Genome Sequencing Improves the Diagnosis of DFNB1 Monoallelic Patients

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Recruitment of Patient/Population
2.3. Whole Genome Sequencing
2.3.1. Laboratory
2.3.2. Bioinformatics
2.4. Data Analysis
2.4.1. SNV Identification
2.4.2. SV Identification
2.5. Confirmation and Segregation Analysis
Variant Confirmation
2.6. Functional Assays Cells
2.6.1. Plasmid Constructs
2.6.2. Mutagenesis
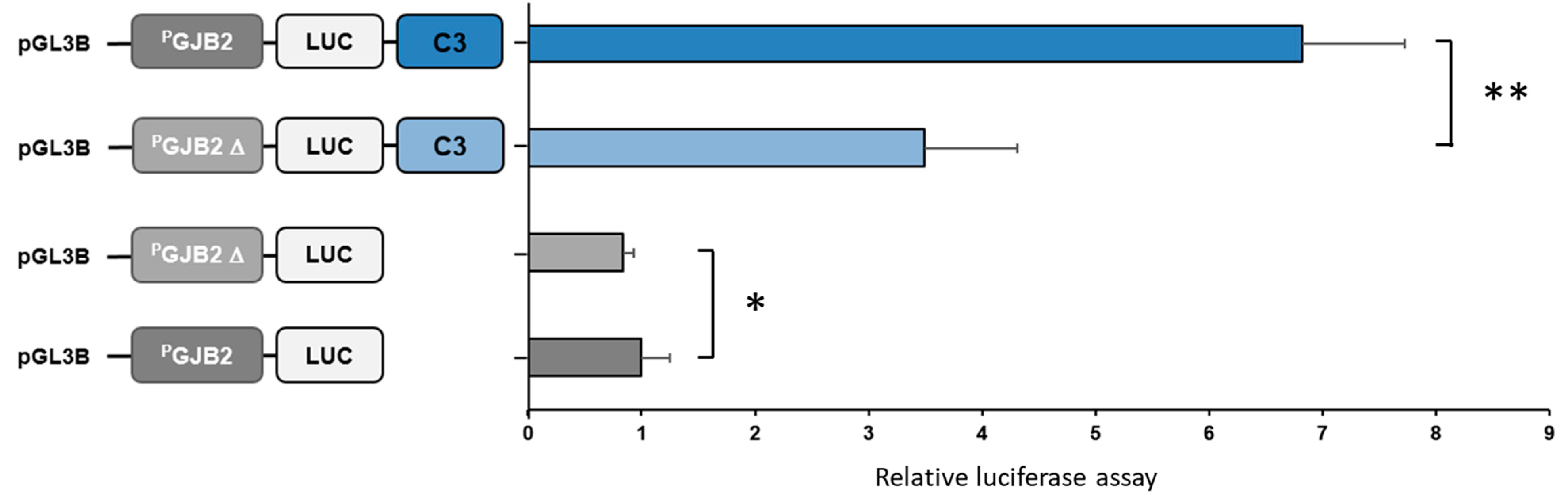
2.6.3. Luciferase Assays
3. Results
3.1. SNV
3.1.1. GJB2 Mutations
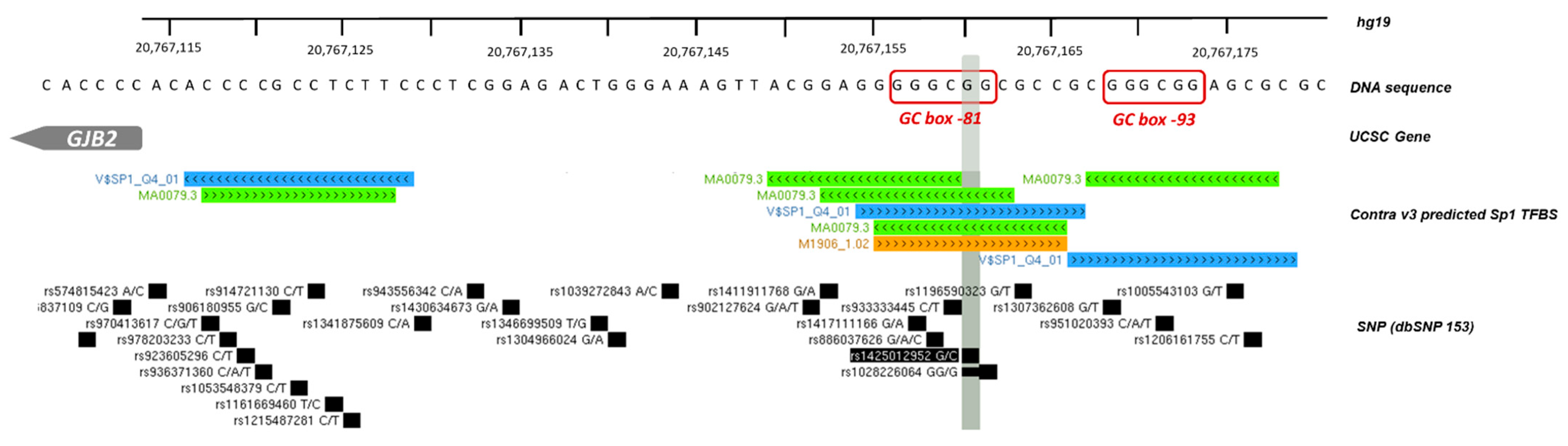
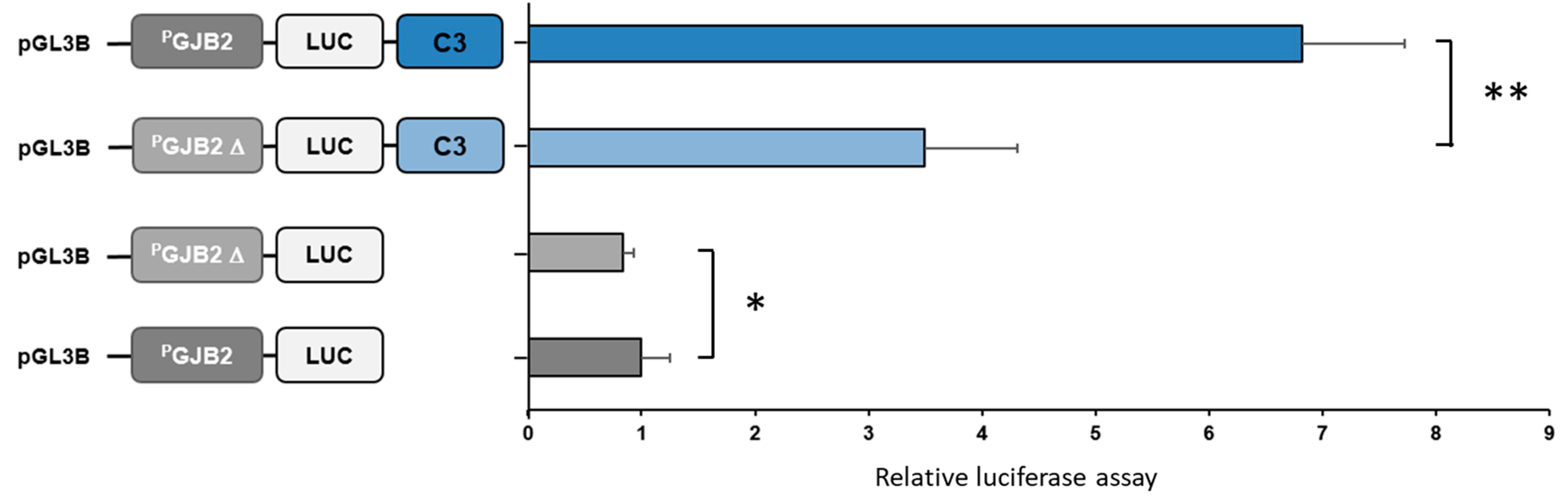
3.1.2. GJB2 Upstream Variation
3.1.3. SNVs on Deafness Genes
USH1C Gene
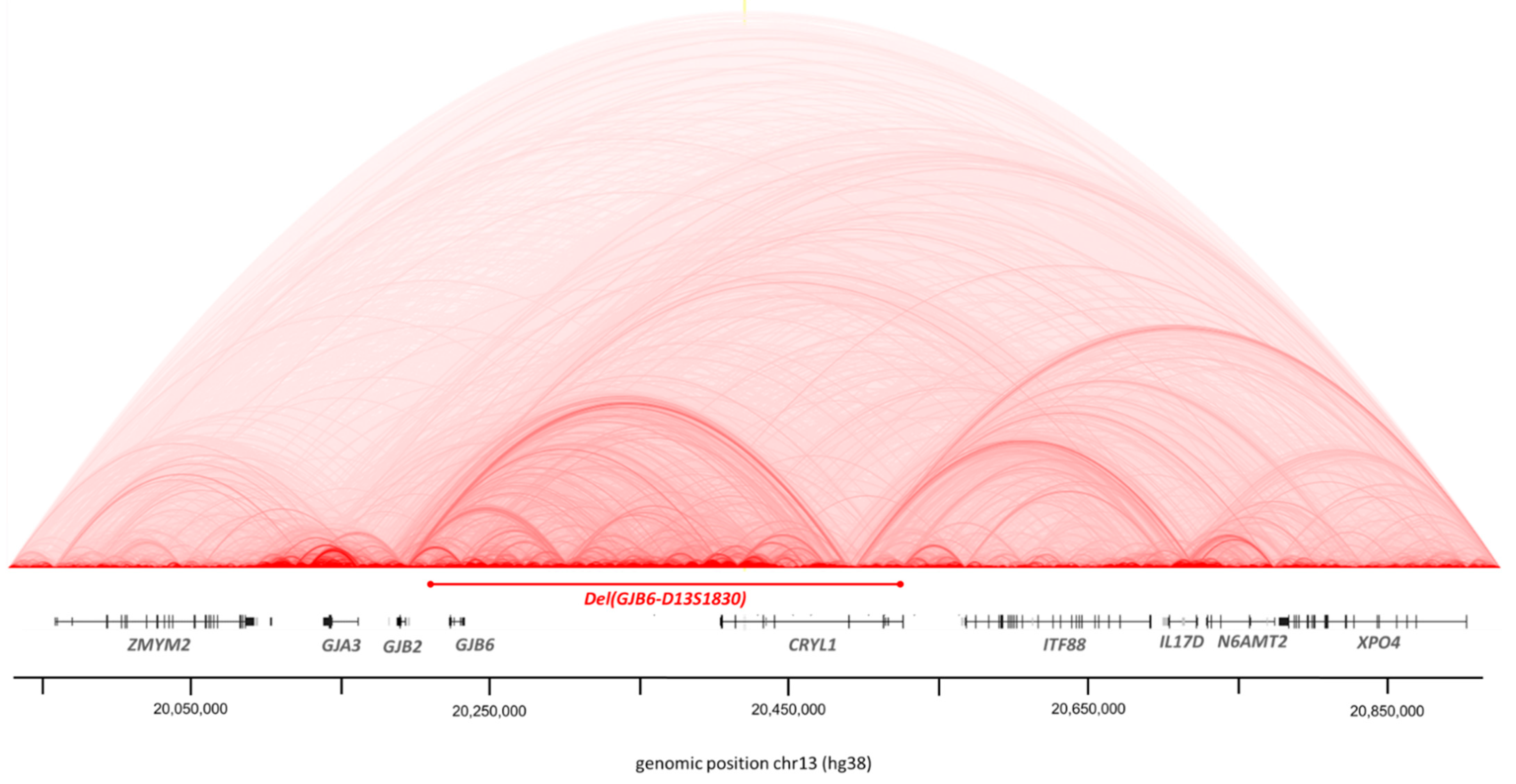
3.2. Structural Variations
3.2.1. CNV by Integragen Genomics
3.2.2. BreakDancer Algorithm for SVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Denoyelle, F.; Marlin, S.; Weil, D.; Moatti, L.; Chauvin, P.; Garabédian, É.-N.; Petit, C. Clinical features of the prevalent form of childhood deafness. Lancet 1999, 353, 1298–1303. [Google Scholar] [CrossRef]
- Feldmann, D.; Denoyelle, F.; Chauvin, P.; Garabédian, E.-N.; Couderc, R.; Odent, S.; Joannard, A.; Schmerber, S.; Delobel, B.; Leman, J.; et al. Large deletion of the GJB6 gene in deaf patients heterozygous for the GJB2 gene mutation: Genotypic and phenotypic analysis. Am. J. Med. Genet. Part A 2004, 127, 263–267. [Google Scholar] [CrossRef]
- Marlin, S.; Feldmann, D.; Blons, H.; Loundon, N.; Rouillon, I.; Albert, S.; Chauvin, P.; Garabédian, E.-N.; Couderc, R.; Odent, S.; et al. GJB2 and GJB6 Mutations: Genotypic and Phenotypic Correlations in a Large Cohort of Hearing-Impaired Patients. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Tanaka-Ouyang, L.; Marlin, S.; Nevoux, J. Les surdités d’origine génétique. Press. Med. 2017, 46, 1089–1096. [Google Scholar] [CrossRef]
- Dror, A.A.; Avraham, K.B. Hearing impairment: A panoply of genes and functions. Neuron 2010, 68, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandía, M.; del Castillo, F.J.; Rodríguez-Álvarez, F.J.; Garrido, G.; Villamar, M.; Calderón, M.; Moreno-Pelayo, M.A.; Moreno, F.; del Castillo, I. A Novel Splice-Site Mutation in the GJB2 Gene Causing Mild Postlingual Hearing Impairment. PLoS ONE 2013, 8, e0073566. [Google Scholar] [CrossRef] [Green Version]
- Leclère, J.-C.; Le Gac, M.-S.; Le Maréchal, C.; Ferec, C.; Marianowski, R. GJB2 mutations: Genotypic and phenotypic correlation in a cohort of 690 hearing-impaired patients, toward a new mutation? Int. J. Pediatr. Otorhinolaryngol. 2017, 102, 80–85. [Google Scholar] [CrossRef]
- Roux, A.-F.; Pallares-Ruiz, N.; Vielle, A.; Faugère, V.; Templin, C.; Leprevost, D.; Artières, F.; Lina, G.; Molinari, N.; Blanchet, P.; et al. Molecular epidemiology of DFNB1 deafness in France. BMC Med. Genet. 2004, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Kenneson, A.; Van Naarden Braun, K.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [Green Version]
- Brozkova, D.S.; Meszarosova, A.U.; Lassuthova, P.; Varga, L.; Staněk, D.; Borecká, S.; Laštůvková, J.; Čejnová, V.; Rašková, D.; Lhota, F.; et al. The Cause of Hereditary Hearing Loss in GJB2 Heterozygotes—A Comprehensive Study of the GJB2/DFNB1 Region. Genes 2021, 12, 684. [Google Scholar] [CrossRef]
- Feldmann, D.; Le Maréchal, C.; Jonard, L.; Thierry, P.; Czajka, C.; Couderc, R.; Ferec, C.; Denoyelle, F.; Marlin, S.; Fellmann, F. A new large deletion in the DFNB1 locus causes nonsyndromic hearing loss. Eur. J. Med. Genet. 2009, 52, 195–200. [Google Scholar] [CrossRef]
- Bliznetz, E.A.; Lalayants, M.R.; Markova, T.G.; Balanovsky, O.P.; Balanovska, E.V.; Skhalyakho, R.A.; Pocheshkhova, E.A.; Nikitina, N.V.; Voronin, S.V.; Kudryashova, E.K.; et al. Update of the GJB2/DFNB1 mutation spectrum in Russia: A founder Ingush mutation del(GJB2-D13S175) is the most frequent among other large deletions. J. Hum. Genet. 2017, 62, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Lerer, I.; Sagi, M.; Ben-Neriah, Z.; Wang, T.; Levi, H.; Abeliovich, D. A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: A novel founder mutation in Ashkenazi Jews. Hum. Mutat. 2001, 18, 460. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, F.J.; Rodríguez-Ballesteros, M.; Alvarez, A.; Hutchin, T.; Leonardi, E.; de Oliveira, C.A.; Azaiez, H.; Brownstein, Z.; Avenarius, M.R.; Marlin, S.; et al. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilch, E.; Azaiez, H.; Fisher, R.A.; Elfenbein, J.; Murgia, A.; Birkenhäger, R.; Bolz, H.; Da Silva-Costa, S.M.; Del Castillo, I.; Haaf, T.; et al. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin. Genet. 2010, 78, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayoun, A.N.A.; Mason-Suares, H.; Frisella, A.L.; Bowser, M.; Duffy, E.; Mahanta, L.; Funke, B.; Rehm, H.L.; Amr, S.S. Targeted Droplet-Digital PCR as a Tool for Novel Deletion Discovery at the DFNB1 Locus. Hum. Mutat. 2015, 37, 119–126. [Google Scholar] [CrossRef]
- Abe, S.; Nishio, S.; Yokota, Y.; Moteki, H.; Kumakawa, K.; Usami, S. Diagnostic pitfalls for GJB2-related hearing loss: A novel deletion detected by Array-CGH analysis in a Japanese patient with congenital profound hearing loss. Clin. Case Rep. 2018, 6, 2111–2116. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, M.; Tome, J.M.; Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. 2020, 21, 292–310. [Google Scholar] [CrossRef]
- Epstein, D.J. Cis-regulatory mutations in human disease. Brief. Funct. Genom. Proteom. 2009, 8, 310–316. [Google Scholar] [CrossRef]
- Kleinjan, D.-J.; Coutinho, P. Cis-ruption mechanisms: Disruption of cis-regulatory control as a cause of human genetic disease. Brief. Funct. Genom. Proteom. 2009, 8, 317–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, F.; Acosta, R.; Addleman, N.J.; Adrian, J.; Afzal, V.; Aken, B.; Akiyama, J.A.; Jammal, O.A.; Amrhein, H.; Anderson, S.M.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 112. [Google Scholar] [CrossRef] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Moisan, S.; Le Nabec, A.; Quillévéré, A.; Le Maréchal, C.; Férec, C. Characterization of GJB2 cis-regulatory elements in the DFNB1 locus. Hum. Genet. 2019, 138, 1275–1286. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stütz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.H.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef]
- Kiang, D.T.; Jin, N.; Tu, Z.J.; Lin, H.H. Upstream genomic sequence of the human connexin 26 gene. Gene 1997, 199, 165–171. [Google Scholar] [CrossRef]
- Tu, Z.J.; Kiang, D.T. Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim. Biophys. Acta 1998, 1443, 169–181. [Google Scholar] [CrossRef]
- Wilch, E.; Zhu, M.; Burkhart, K.B.; Regier, M.; Elfenbein, J.L.; Fisher, R.A.; Friderici, K.H. Expression of GJB2 and GJB6 is reduced in a novel DFNB1 allele. Am. J. Hum. Genet. 2006, 79, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Matos, T.D.; Caria, H.; Simões-Teixeira, H.; Aasen, T.; Nickel, R.; Jagger, D.J.; O’Neill, A.; Kelsell, D.P.; Fialho, G. A novel hearing loss-related mutation occurring in the GJB2 basal promoter. J. Med. Genet. 2007, 44, 721–725. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Paris, J.; Ballay, C.; Inserra, M.; Stidham, K.; Colen, T.; Roberson, J.; Gardner, P.; Schrijver, I. Genetic analysis of presbycusis by arrayed primer extension. Ann. Clin. Lab. Sci. 2008, 38, 352–360. [Google Scholar]
- Fetoni, A.R.; Zorzi, V.; Paciello, F.; Ziraldo, G.; Peres, C.; Raspa, M.; Scavizzi, F.; Salvatore, A.M.; Crispino, G.; Tognola, G.; et al. Cx26 partial loss causes accelerated presbycusis by redox imbalance and dysregulation of Nfr2 pathway. Redox Biol. 2018, 19, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. bioRxiv 2021, 445798. [Google Scholar] [CrossRef]
- Vollger, M.R.; Guitart, X.; Dishuck, P.C.; Mercuri, L.; Harvey, W.T.; Gershman, A.; Diekhans, M.; Sulovari, A.; Munson, K.M.; Lewis, A.M.; et al. Segmental duplications and their variation in a complete human genome. bioRxiv 2021, 445678. [Google Scholar] [CrossRef]
- Fan, X.; Abbott, T.E.; Larson, D.; Chen, K. BreakDancer: Identification of genomic structural variation from paired-end read mapping. Curr. Protoc. Bioinform. 2014, 45, 15.6.1–15.6.11. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, T.; Lee, W.P.; Leone, J.; Zhu, Q.; Zhang, C.; Liu, S.; Sargent, J.; Shanker, K.; Mil-homens, A.; Cerveira, E.; et al. FusorSV: An algorithm for optimally combining data from multiple structural variation detection methods. Genome Biol. 2018, 19, 38. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Deafness Phenotype | Case | Gene | HGVSc | Chr. | Position (hg38) | HGVSp | Impact | Consequences | Variant Class | rs Number | Allele Frequency (GnomAD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | Profound | Single | GJB2 | NM_004004.5:c.132G > A | chr13 | 20189450 | NP_003995.2:p.Trp44Ter | HIGH | stop_gained | SNV | rs104894407 | 0.00001470 |
| P2 | Profound | Single | GJB2 | NM_004004.5:c.169C > T | chr13 | 20189413 | NP_003995.2:p.Gln57Ter | HIGH | stop_gained | SNV | rs111033297 | 0.00005879 |
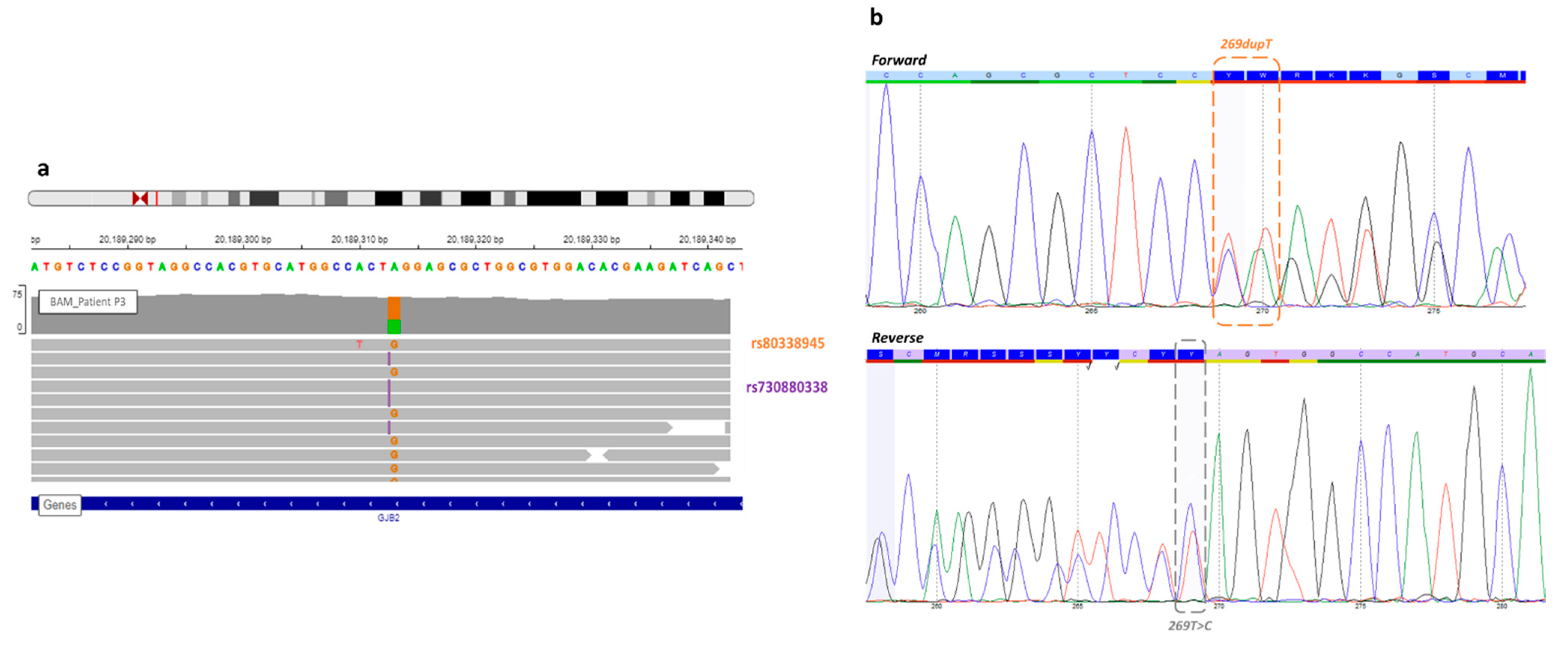
| P3 | Profound | Single | GJB2 | NM_004004.5:c.269dup | chr13 | 20189312 | NP_003995.2:p.Val91SerfsTer11 | HIGH | frameshift_variant | insertion | rs730880338 | 0.00002940 |
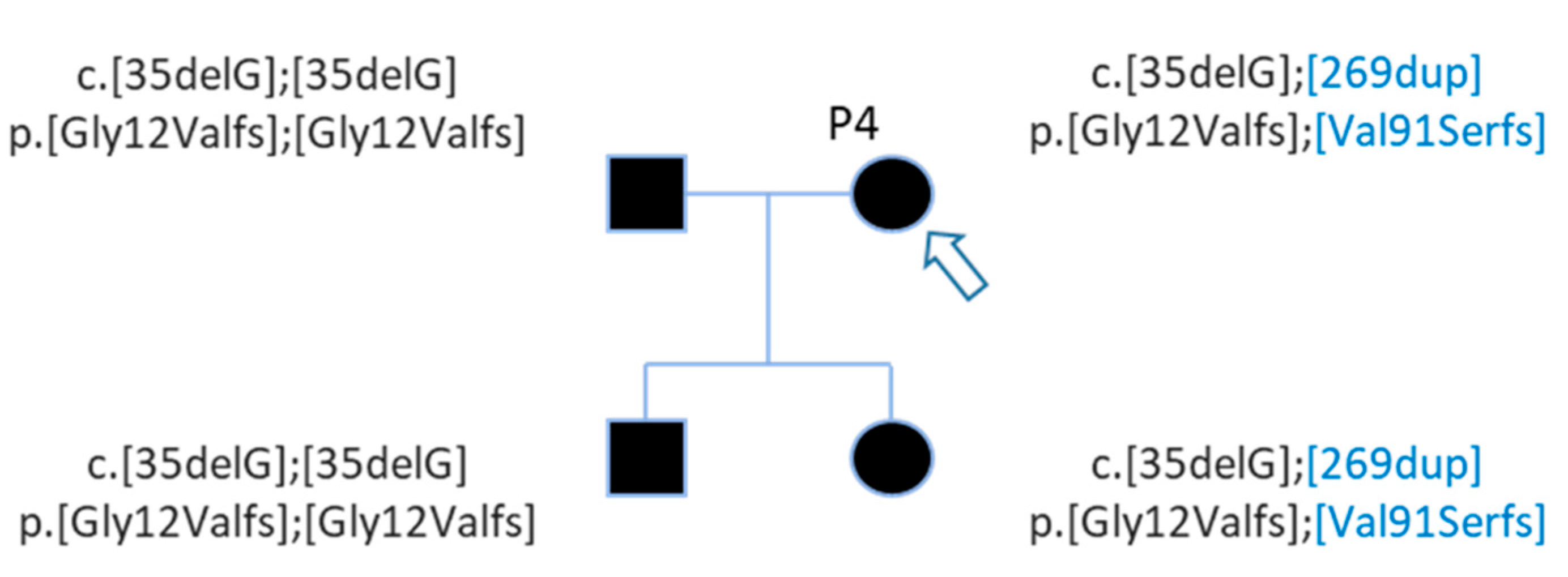
| P4 | Profound | Family | GJB2 | NM_004004.5:c.35del | chr13 | 20189546 | NP_003995.2:p.Gly12ValfsTer2 | HIGH | frameshift_variant | deletion | rs80338939 | 0.009802 |
| P5 | Profound | Single | del(GJB6-D13S1830) | 0.0003935 | ||||||||
| P6 | Profound | Single | GJB2 | NM_004004.5:c.169C > T | chr13 | 20189413 | NP_003995.2:p.Gln57Ter | HIGH | stop_gained | SNV | rs111033297 | 0.00005879 |
| P7 | Profound | Single | GJB2 | NM_004004.5:c.633T > A | chr13 | 20188949 | NP_003995.2:p.Cys211Ter | HIGH | stop_gained | SNV | - | - |
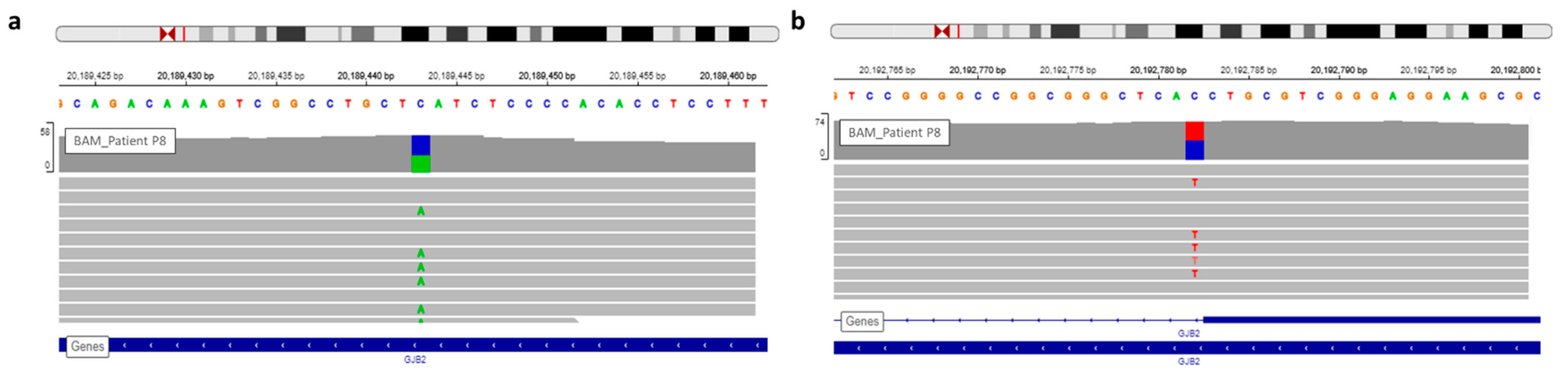
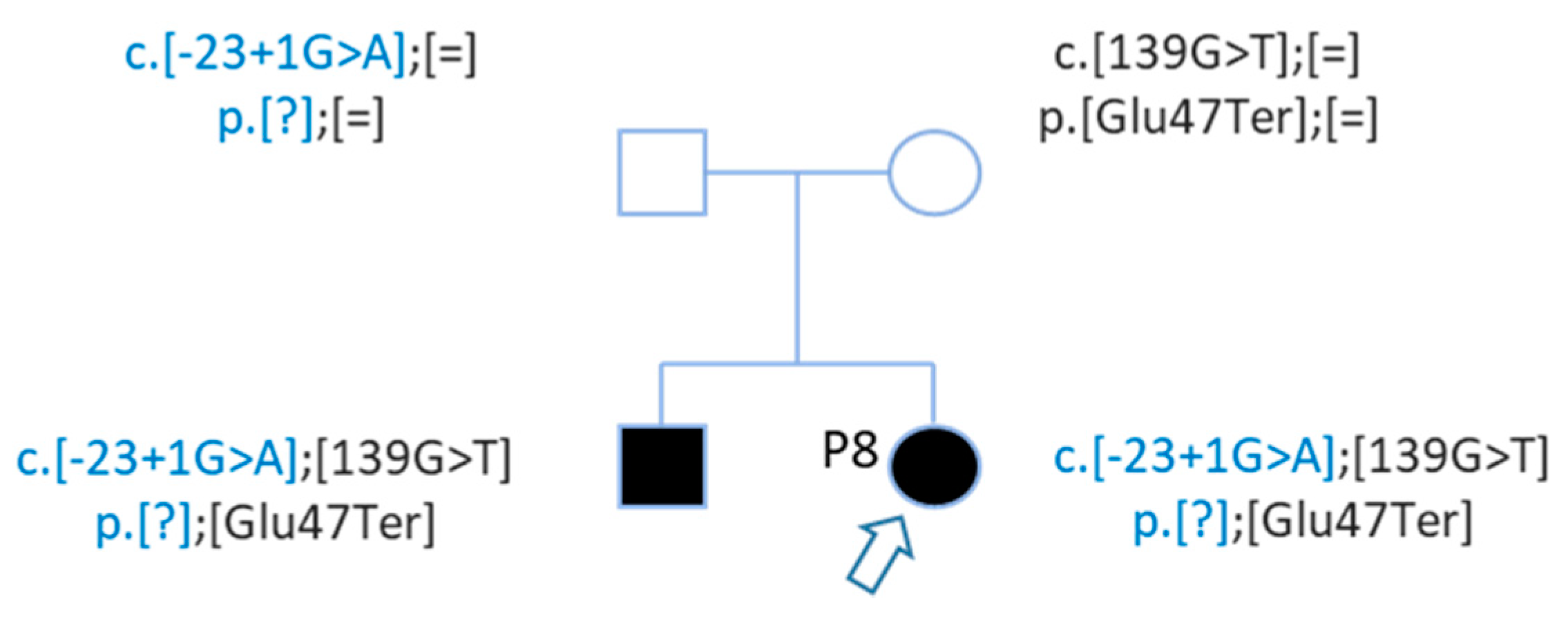
| P8 | Profound | Family | GJB2 | NM_004004.5:c.139G > T | chr13 | 20189443 | NP_003995.2:p.Glu47Ter | HIGH | stop_gained | SNV | rs104894398 | 0.0001176 |
| P9 | Profound | Single | GJB2 | NM_004004.5:c.313_326del | chr13 | 20189255 | NP_003995.2:p.Lys105GlyfsTer5 | HIGH | frameshift_variant | deletion | rs111033253 | 0.0003234 |
| P10 | Mild | Single | GJB2 | NM_004004.5:c.139G > T | chr13 | 20189443 | NP_003995.2:p.Glu47Ter | HIGH | stop_gained | SNV | rs104894398 | 0.0001176 |
| Patient | Gene | HGVSc | Chr. | Position (hg38) | HGVSp | Impact | Consequences | Variant Class | rs Number | Allele Frequency (GnomAD) |
|---|---|---|---|---|---|---|---|---|---|---|
| P3 | GJB2 | NM_004004.5:c.269dup | chr13 | 20189312 | NP_003995.2:p.Val91SerfsTer11 | HIGH | frameshift_variant | insertion | rs730880338 | 0.00002940 |
| GJB2 | NM_004004.5:c.269T > C | chr13 | 20189313 | NP_003995.2:p.Leu90Pro | MODERATE | missense_variant | SNV | rs80338945 | 0.001161 | |
| P4 | GJB2 | NM_004004.5:c.35del | chr13 | 20189546 | NP_003995.2:p.Gly12ValfsTer2 | HIGH | frameshift_variant | deletion | rs80338939 | 0.009802 |
| GJB2 | NM_004004.5:c.269dup | chr13 | 20189312 | NP_003995.2:p.Val91SerfsTer11 | HIGH | frameshift_variant | insertion | rs730880338 | 0.00002940 | |
| P8 | GJB2 | NM_004004.5:c.139G > T | chr13 | 20189443 | NP_003995.2:p.Glu47Ter | HIGH | stop_gained | SNV | rs104894398 | 0.0001176 |
| GJB2 | NM_004004.5:c.-23 + 1G > A | chr13 | 20192782 | . | HIGH | splice_donor_variant | SNV | rs80338940 | 0.0003236 | |
| P10 | GJB2 | NM_004004.5:c.139G > T | chr13 | 20189443 | NP_003995.2:p.Glu47Ter | HIGH | stop_gained | SNV | rs104894398 | 0.0001176 |
| GJB2 | G > C | chr13 | 20193022 | - | UNKNOWN | upstream_gene_variant | SNV | rs1425012952 | 0.00001472 | |
| GJB2 | G > T | chr13 | 20183294 | - | UNKNOWN | downstream_gene_variant | SNV | rs372782198 | 0.003099 |
| Gene | HGVSc | Chr. | Position (hg38) | Protein Variation | Impact | Consequences | Variant Class | rs Number | Allele Frequency (GnomAD) |
|---|---|---|---|---|---|---|---|---|---|
| USH1C | NM_001297764.1:c.463C > T | chr11 | 17527256 | R155 * | HIGH | stop_gained | SNV | rs377145777 | 0.00001759 |
| USH1C | NM_001297764.1:c.1589 + 3_1589 + 6del | chr11 | 17496751 | ... | LOW | splice_region_variant&intron_variant | deletion | - | - |
| USH1C | NM_005709.3:c.1557G > C | chr11 | 17498195 | E519D | MODERATE | missense_variant | SNV | rs1064074 | 0.5511 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Nabec, A.; Collobert, M.; Le Maréchal, C.; Marianowski, R.; Férec, C.; Moisan, S. Whole-Genome Sequencing Improves the Diagnosis of DFNB1 Monoallelic Patients. Genes 2021, 12, 1267. https://doi.org/10.3390/genes12081267
Le Nabec A, Collobert M, Le Maréchal C, Marianowski R, Férec C, Moisan S. Whole-Genome Sequencing Improves the Diagnosis of DFNB1 Monoallelic Patients. Genes. 2021; 12(8):1267. https://doi.org/10.3390/genes12081267
Chicago/Turabian StyleLe Nabec, Anaïs, Mégane Collobert, Cédric Le Maréchal, Rémi Marianowski, Claude Férec, and Stéphanie Moisan. 2021. "Whole-Genome Sequencing Improves the Diagnosis of DFNB1 Monoallelic Patients" Genes 12, no. 8: 1267. https://doi.org/10.3390/genes12081267
APA StyleLe Nabec, A., Collobert, M., Le Maréchal, C., Marianowski, R., Férec, C., & Moisan, S. (2021). Whole-Genome Sequencing Improves the Diagnosis of DFNB1 Monoallelic Patients. Genes, 12(8), 1267. https://doi.org/10.3390/genes12081267