
Identification of New Chromosomal Loci Involved in com Genes Expression and Natural Transformation in the Actinobacterial Model Organism Micrococcus luteus

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. EMS Mutagenesis and Mutagenesis Efficiency Assessment
2.3. Linkage Analysis
2.4. β-Galactosidase Activity Assays
2.5. Genomic DNA Extraction from M. luteus
2.6. Genome Sequencing, Assembly, Quality Analysis, and Mutations Plotting
2.7. SNP Clean Insertions and Construction of Gene Deletion Mutants
2.8. Extraction of Total RNA, 5′RACE and 5′UTR Analyses
2.9. Transformation Frequency Assay
3. Results
3.1. Chemical Mutagenesis
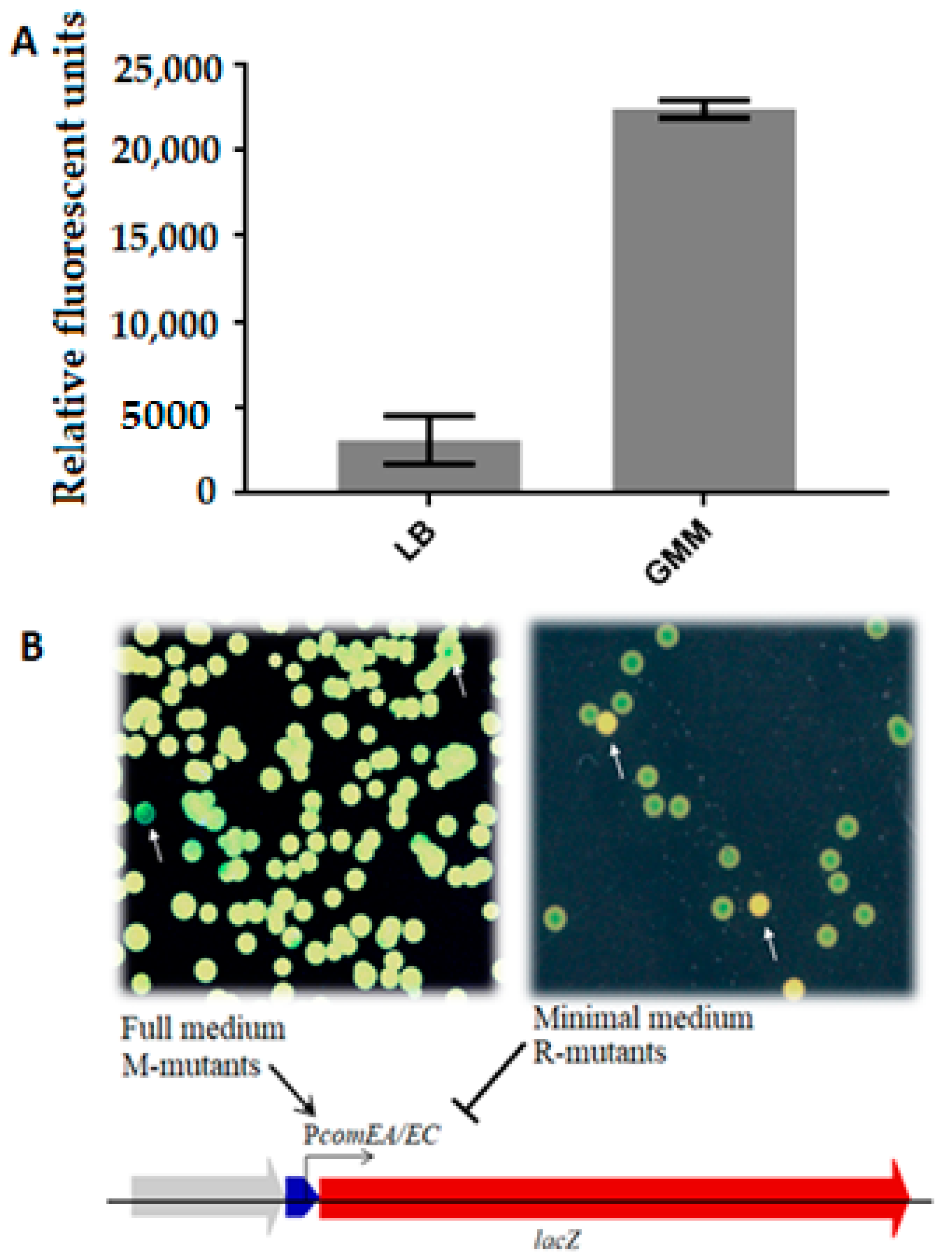
3.2. Mutant Screening and Phenotype Selection
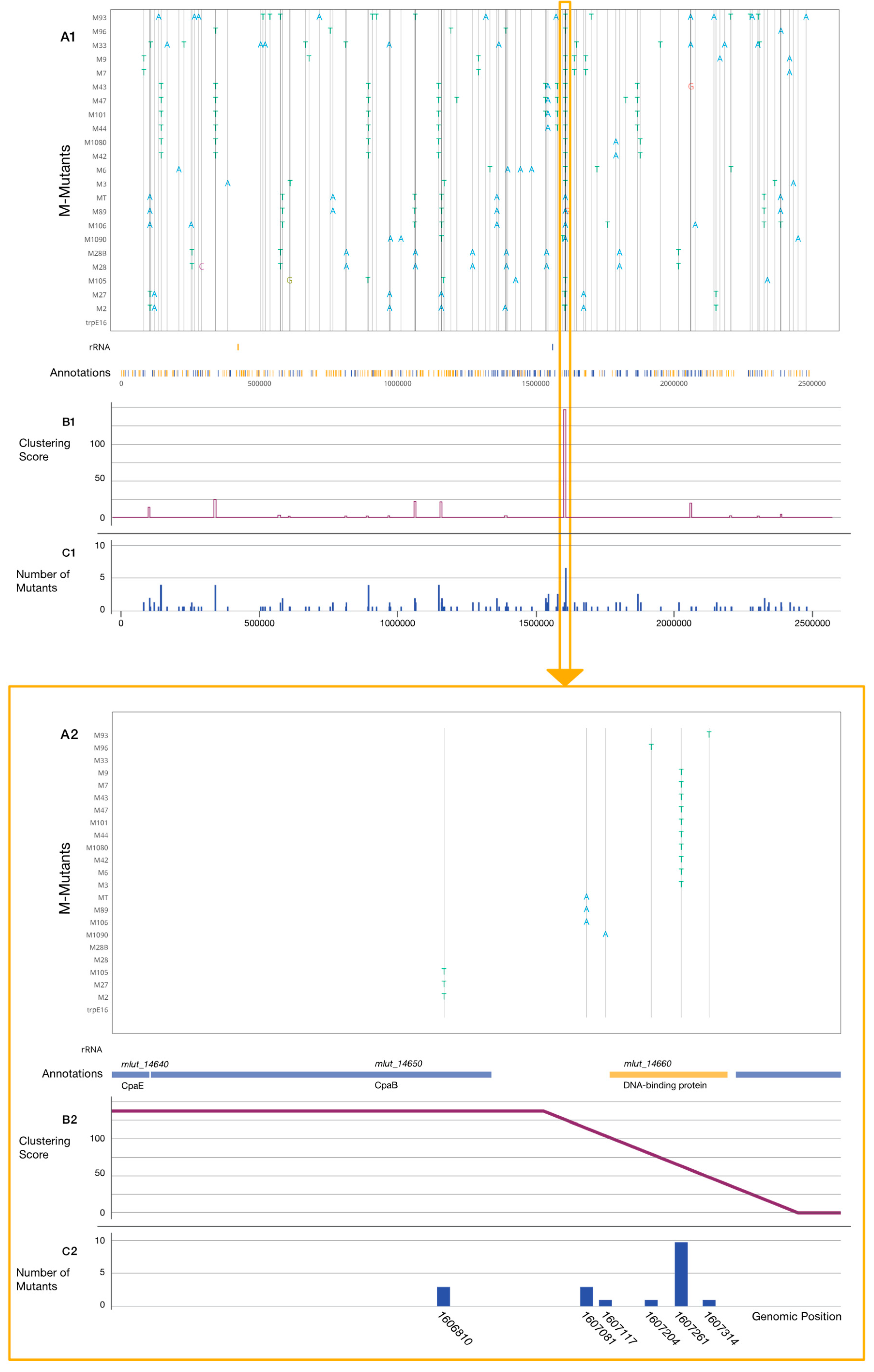
3.3. Genome Sequencing and Mutation Analysis
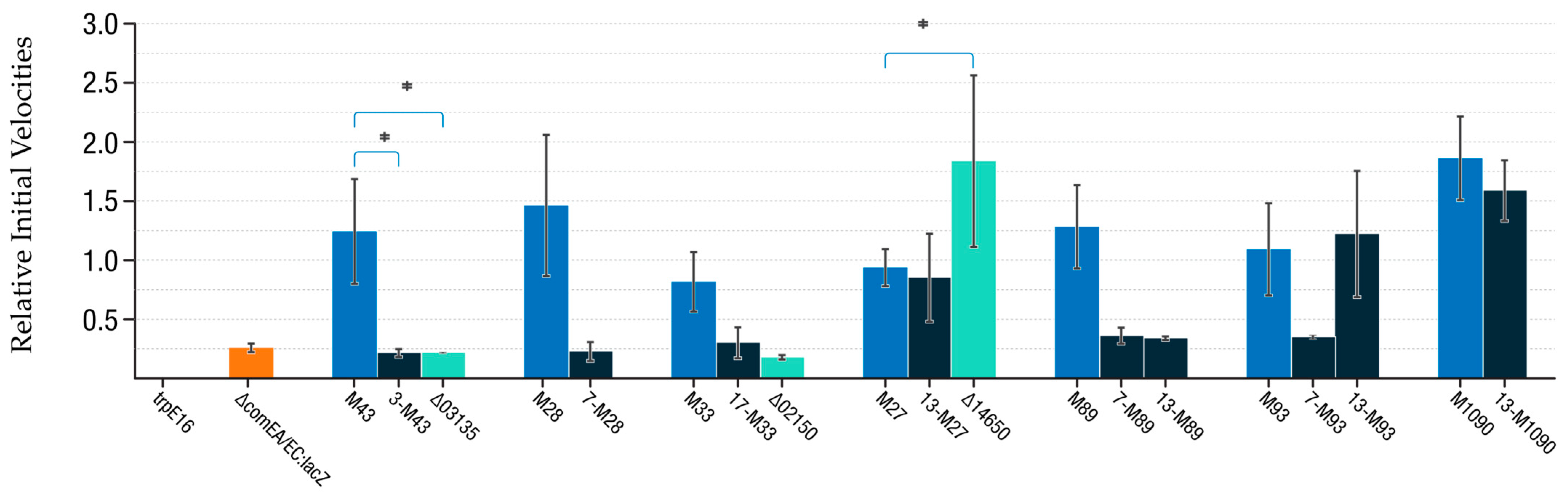
3.4. Introduction of SNPs and Gene Deletions into M. luteus and Evaluation of comEA/EC Promoter Activity
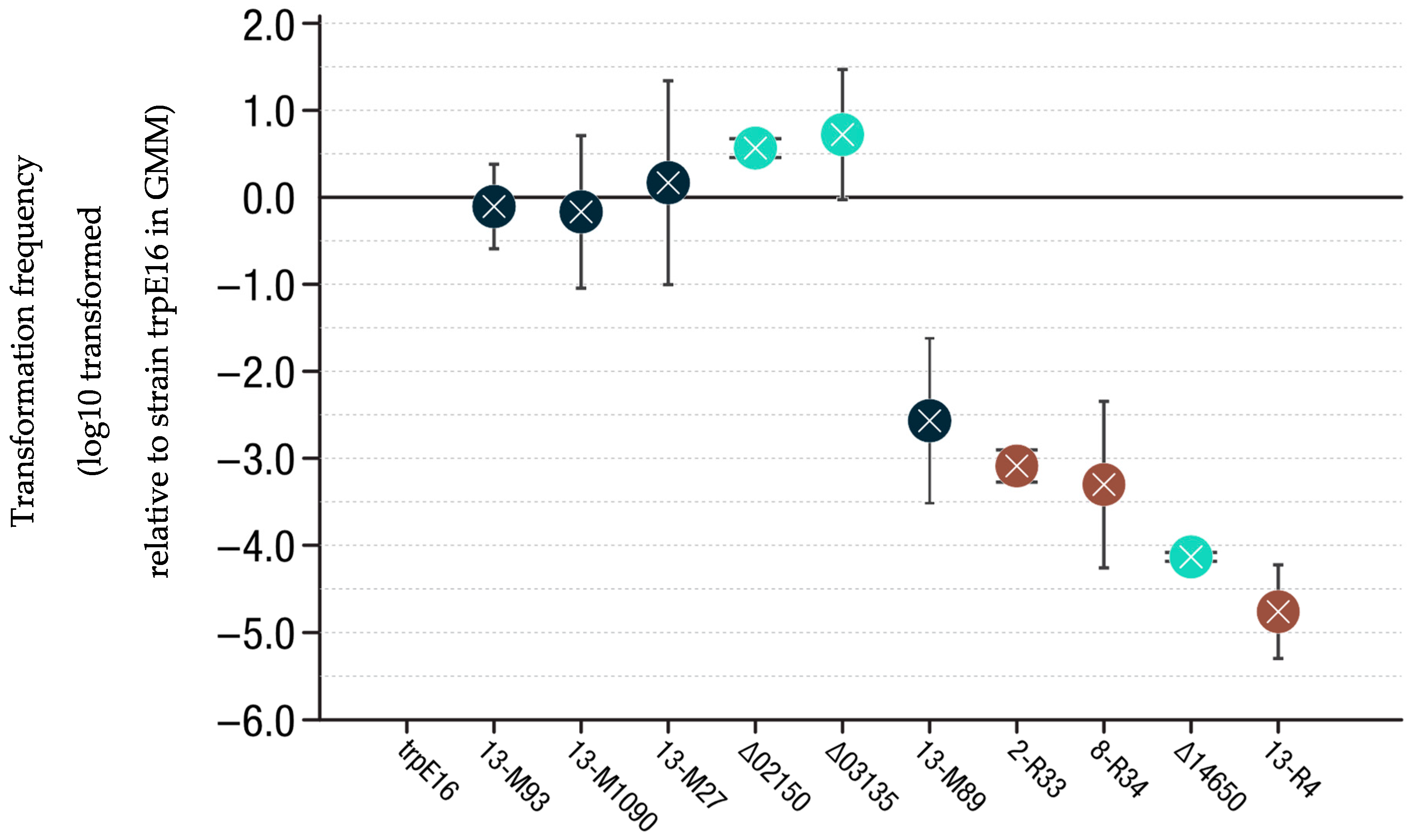
3.5. Transformation Frequency Evaluation
3.6. Genomic Synteny of Cluster 13-Associated Genes
4. Discussion
4.1. Mutant Selection and Clustering of Mutations
4.2. R-Mutations
4.3. M-Mutations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yue, J.; Hu, X.; Sun, H. Widespread impact of horizontal gene transfer on plant colonization of land. Nat. Commun. 2012, 3, 1152–1159. [Google Scholar] [CrossRef] [Green Version]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Peter Gogarten, J.; Townsend, J.P. Horizontal gene transfer accelerates genome innovation and evolution. Mol. Biol. Evol. 2005, 20, 1598–1602. [Google Scholar]
- Johnsborg, O.; Eldholm, V.; Håvarstein, L.S. Natural genetic transformation: Prevalence, mechanisms and function. Res. Microbiol. 2007, 158, 767–778. [Google Scholar] [CrossRef]
- Johnston, C.; Martin, B.; Fichant, G. Bacterial transformation: Distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 2014, 12, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Griffith, F. The Significance of Pneumococcal Types. J. Hyg. 1928, 27, 113–159. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, H.L.; Dillard, J.P. Natural transformation of Neisseria gonorrhoeae: From DNA donation to homologous recombination. Mol. Microbiol. 2006, 59, 376–385. [Google Scholar] [CrossRef]
- Maughan, H.; Sinha, S.; Wilson, L. Competence, DNA Uptake, and Transformation in Pasteurellaceae. Pasteurellaceae Biol. Genomics Mol. Asp. 2008, 79–98. [Google Scholar]
- Matthey, N.; Blokesch, M. The DNA-Uptake Process of Naturally Competent Vibrio cholerae. Trends. Microbiol. 2016, 24, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Dubnau, D. DNA Uptake during bacterial transformation. Nat. Rev. Microbiol 2004, 2, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Averhoff, B. Shuffling genes around in hot environments: The unique DNA transporter of Thermus thermophilus. FEMS. Microbiol. Rev. 2009, 33, 611–626. [Google Scholar] [CrossRef] [Green Version]
- Dubnau, D. The regulation of genetic competence in Bacillus subtilis. Mol. Microbiol. 1991, 5, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Johnsborg, O.; Håvarstein, L.S. Regulation of natural genetic transformation and acquisition of transforming DNA in Streptococcus pneumoniae, FEMS. Microbiol. Rev. 2009, 33, 627–642. [Google Scholar]
- Kloos, W.E. Factors affecting transformation of Micrococcus lysodeikticus. J. Bacteriol. 1969, 98, 1397–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloos, W.E.; Schultes, L.M. Transformation in Micrococcus lysodeikticus. J. Gen. Microbiol. 1969, 55, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelov, A.; Bergen, P.; Nadler, F.; Hornburg, P.; Lichev, A.; Überlacker, M.; Pachl, F.; Kuster, B.; Liebl, W. Novel Flp pilus biogenesis-dependent natural transformation. Front Microbiol. 2015, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Inamine, G.S.; Dubnau, D. ComEA, a Bacillus subtilis integral membrane protein required for genetic transformation, is needed for both DNA binding and transport. J. Bacteriol. 1995, 177, 3045–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provvedi, R.; Dubnau, D. ComEA is a DNA receptor for transformation of competent Bacillus subtilis. Mol. Microbiol. 1999, 31, 271–280. [Google Scholar] [CrossRef]
- Claverys, J.P.; Prudhomme, M.; Martin, B. Induction of competence regulons as a general response to stress in gram-positive bacteria. Annu. Rev. Microbiol. 2006, 60, 451–475. [Google Scholar] [CrossRef]
- Shanker, E.; Federle, M.J. Quorum Sensing Regulation of Competence and Bacteriocins in Streptococcus pneumoniae and mutans. Genes 2017, 8, 15. [Google Scholar] [CrossRef]
- Lee, M.S.; Morrison, D.A. Identification of a New Regulator in Streptococcus pneumoniae Linking Quorum Sensing to Competence for Genetic Transformation. J. Bacteriol. 1999, 181, 5004–5016. [Google Scholar] [CrossRef]
- Mohan, S.; Dubnau, D. Transcriptional Regulation of comC: Evidence for a Competence-Specific Transcription Factor in Bacillus subtilis. Am. Soc. Microbiol. 1990, 172, 4064–4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maamar, H.; Dubnau, D. Bistability in the Bacillus subtilis K-state (competence) system requires a positive feedback loop. Mol. Microbiol. 2005, 56, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leisner, M.; Stingl, K.; Frey, E.; Maier, B. Stochastic switching to competence. Curr. Opin. Microbiol. 2008, 11, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Jaskólska, M.; Stutzmann, S.; Stoudmann, C.; Blokesch, M. QstR-dependent regulation of natural competence and type VI secretion in Vibrio. Cholerae. Nucleic Acids Res. 2018, 46, 10619–10634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichev, A.; Angelov, A.; Cucurull, I.; Liebl, W. Amino acids as nutritional factors and (p)ppGpp as an alarmone of the stringent response regulate natural transformation in Micrococcus luteus. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Dubnau, D.; Roggiani, M. Growth Medium-Independent Genetic Competence Mutants of Bacillus subtilis. J. Bacteriol. 1990, 172, 4048–4055. [Google Scholar] [CrossRef] [Green Version]
- Shields, R.C.; O’Brien, G.; Maricic, N.; Kesterson, A.; Grace, M.; Burne, R.A.; Hagen, S.J. Genome-Wide Screens Reveal New Gene Products That Influence Genetic Competence in Streptococcus mutans. J Bacteriol. 2018, 200, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kloos, W.E.; Rose, N.E. Transformation mapping of tryptophan loci in Micrococcus luteus. Genetics 1970, 66, 595–605. [Google Scholar] [CrossRef]
- Lennox, E.S. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1955, 1, 190–206. [Google Scholar] [CrossRef]
- Wolin, H.L.; Naylor, H.B. Basic nutritional requirements of Micrococcus lysodeikticus. J. Bacteriol. 1957, 74, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Baltz, R.H. Spontaneous and induced mutations to rifampicin, streptomycin and spectinomycin resistances in actinomycetes: Mutagenic mechanisms and applications for strain improvement. J. Antibiot. 2014, 67, 619–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef] [Green Version]
- Kostner, D.; Peters, B.; Mientus, M.; Liebl, W.; Ehrenreich, A. Importance of codB for new codA-based markerless gene deletion in Gluconobacter strains. Appl. Microbiol. Biotechnol. 2013, 97, 8341–8349. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Neuhard, J. Utilization of preformed pyrimidine bases and nucleosides. In Metabolims of Nucleotides, Nucleosides and Nucleobases in Microorganisms; Munch-Petersen, A., Ed.; Acad Press: New York, NY, USA, 1983; pp. 95–148. [Google Scholar]
- Martinussen., J.; Hammer, K. Cloning and characterization of upp, a gene encoding uracil phosphoribosyltransferase from Lactococcus lactis. J. Bacteriol. 1994, 176, 6457–6463. [Google Scholar] [CrossRef] [Green Version]
- Sega, G.A. A review of the genetic effects of ethyl methanesulfonate. Mutat. Res. Genet. Toxicol. 1984, 134, 113–142. [Google Scholar] [CrossRef]
- Perkins, J.B.; Youngman, P.J. Construction and properties of Tn917-lac, a transposon derivative that mediates transcriptional gene fusions in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 1986, 83, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Hahn, J.; Albano, M.; Dubnau, D. Isolation and characterization of Tn917lac-Generated competence mutants of Bacillus subtilis. J. Bacteriol. 1987, 169, 4469–4478. [Google Scholar] [CrossRef] [Green Version]
- Albano, M.; Hahn, J.; Dubnau, D. Expression of competence genes in Bacillus subtilis. J. Bacteriol. 1987, 169, 3110–3117. [Google Scholar] [CrossRef] [Green Version]
- Turgay, K.; Hahn, J.; Burghoorn, J.; Dubnau, D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998, 17, 6730–6738. [Google Scholar] [PubMed] [Green Version]
- Morton, N.E. Sequential tests for the detection of linkage. Am. J. Hum. Genet. 1955, 7, 277–318. [Google Scholar]
- Batut, B.; Hiltemann, S.; Bagnacani, A.; Baker, D.; Bhardwaj, V.; Blank, C.; Bretaudeau, A.; Brillet-Guéguen, L.; Ceck, M.; Chilton, J.; et al. Community-Driven Data Analysis Training for Report Community-Driven Data Analysis Training for Biology. Cell Syst. 2018, 6, 752–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotin, S.; Fuller, J.D.; Bast, D.J.; Beveridge, T.J.; de Azavedo, J.C. Capsule expression regulated by a two-component signal transduction system in Streptococcus iniae, FEMS. Immunol. Med. Microbiol. 2007, 50, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, L.M.; Aravind, L. The emergence of catalytic and structural diversity within the β-clip fold. Proteins Struct. Funct. Genet. 2004, 55, 977–991. [Google Scholar] [CrossRef]
- Crozat, E.; Rousseau, P.; Fournes, F.; Cornet, F. The FtsK Family of DNA Translocases Finds the Ends of Circles. J. Mol. Microbiol. Biotechnol. 2014, 24, 396–408. [Google Scholar] [CrossRef]
- Barre, F. FtsK and SpoIIIE: The tale of the conserved tails. Mol. Microbiol. 2007, 66, 1051–1055. [Google Scholar] [CrossRef]
- Koonin, E.V. A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids. Res. 1993, 21, 2541–2547. [Google Scholar] [CrossRef]
- Foulger, D.; Errington, J. The role of the sporulation gene spolllE in the regulation of prespore-specific gene expression in Bacillus subtilis. Mol. Microbiol. 1989, 3, 1247–1255. [Google Scholar] [CrossRef]
- Pelton, J.G.; Kustu, S.; Wemmer, D.E. Solution Structure of the DNA-binding Domain of NtrC with Three Alanine Substitutions. J. Mol. Biol. 1999, 292, 1095–1110. [Google Scholar] [CrossRef]
- Bickle, T.A.; Kruger, D.H. Biology of DNA Restriction. Microbiol. Rev. 1993, 57, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Wada, C.; Kawagoe, Y.; Ara, T.; Maeda, M.; Masuda, Y.; Hiraga, S.; Mori, H. Genome-wide analysis of deoxyadenosine methyltransferase-mediated control of gene expression in Escherichia coli. Mol. Microbiol. 2002, 45, 673–695. [Google Scholar] [CrossRef]
- Baker, T.; Sauer, R. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Espah Borujeni, A.; Salis, H.M. Translation Initiation is Controlled by RNA Folding Kinetics via a Ribosome Drafting Mechanism. J. Am. Chem. Soc. 2016, 138, 7016–7023. [Google Scholar] [CrossRef]
- Tomich, M.; Planet, P.J.; Figurski, D.H. The tad locus: Postcards from the widespread colonization island. Nat. Rev. Microbiol. 2007, 5, 363–375. [Google Scholar] [CrossRef]
- Clock, S.A.; Planet, P.J.; Perez, B.A.; Figurski, D.H. Outer Membrane Components of the Tad (Tight Adherence) Secreton of Aggregatibacter actinomycetemcomitans. J. Bacteriol. 2008, 190, 980–990. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Christen, B.; Chiu, H.J.; Jaroszewski, L.; Klock, H.E.; Knuth, M.W.; Miller, M.D.; Elsliger, M.A.; Deacon, A.M.; Godzik, A.; et al. Structure of the pilus assembly protein TadZ from Eubacterium rectale: Implications for polar localization. Mol. Microbiol. 2012, 83, 712–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-cheeks, B.A.; Planet, P.J.; Sarkar, I.N.; Clock, S.A.; Xu, Q.; Figurski, D.H. The product of tadZ, a new member of the parA/minD superfamily, localizes to a pole in Aggregatibacter actinomycetemcomitans. Mol. Microbiol. 2012, 83, 694–711. [Google Scholar] [CrossRef] [Green Version]
- Möglich, A.; Ayers, R.; Moffat, K. Structure and Signaling Mechanism of Per-ARNT-Sim Domains. Structure 2009, 17, 1282–1294. [Google Scholar] [CrossRef] [Green Version]
- Bernard, C.S.; Bordi, C.; Termine, E.; Filloux, A.; de Bentzmann, S. Organization and PprB-Dependent Control of the Pseudomonas aeruginosa tad Locus, Involved in Flp Pilus Biology. J. Bacteriol. 2009, 191, 1961–1973. [Google Scholar] [CrossRef] [Green Version]
- Viollier, P.H.; Sternheim, N.; Shapiro, L. A dynamically localized histidine kinase controls the asymmetric distribution of polar pili proteins. EMBO J. 2002, 21, 4420–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viollier, P.H.; Sternheim, N.; Shapiro, L. Identification of a localization factor for the polar positioning of bacterial structural and regulatory proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 13831–13836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felder, Y. Analysis of Biological Networks: Transcriptional Networks-Promoter Sequence Analysis. Gene 2007, 1–15. [Google Scholar]
- Shearwin, K.E.; Callen, B.P.; Egan, J.B. Transcriptional interference-A crash course. Trends. Genet. 2005, 21, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Yu, Y.; Wang, L.; Luo, Y.; Guo, J.H.; Chai, Y. The comER gene plays an important role in biofilm formation and sporulation in both Bacillus subtilis and Bacillus cereus. Front. Microbiol. 2016, 7, 1–16. [Google Scholar] [CrossRef]
- She, Q.; Hunter, E.; Qin, Y.; Nicolau, S.; Zalis, E.A.; Wang, H.; Chen, Y.; Chai, Y. Negative interplay between biofilm formation and competence in the environmental strains of Bacillus subtilis. bioRxiv 2020, 5, 17–20. [Google Scholar] [CrossRef]
- Surger, M.J.; Angelov, A.; Stier, P.; Übelacker, M.; Liebl, W. Impact of branched-chain amino acid catabolism on fatty acid and alkene biosynthesis in Micrococcus luteus. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]

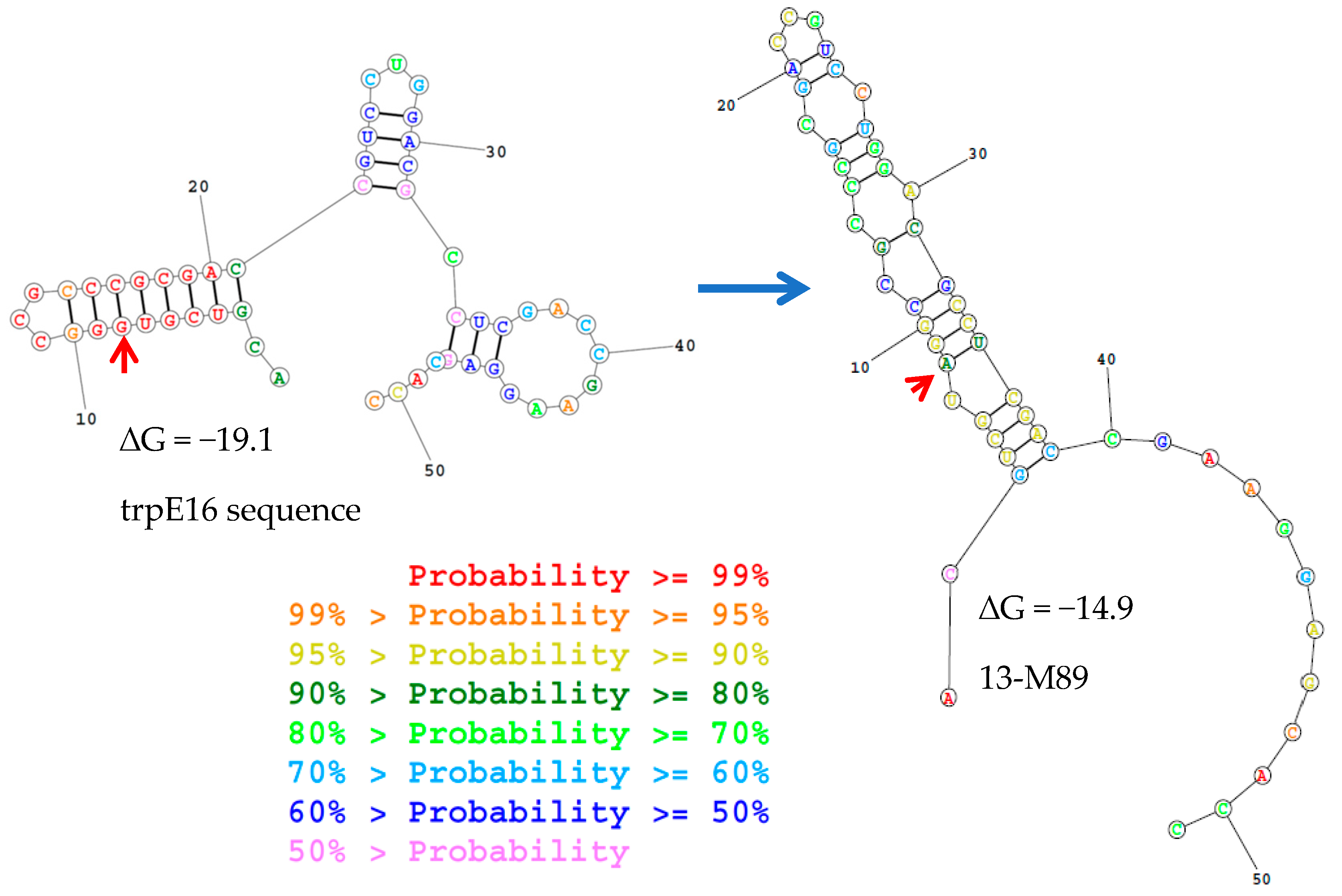


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Genotype and Relevant Phenotype | Source |
|---|---|---|
| trpE16 | trpE16, mutagenesis derivative of ATCC 27141, Trp− | [29] |
| ΔcomEA/EC:lacZ | trpE16 ΔcomEA/EC:lacZ-Kan;KanR, LacZ expression | [26] |
| ∆14650 | trpE16 ∆Mlut_14650 | This study |
| ∆02150 | trpE16 ∆Mlut_02150 | This study |
| ∆03135 | trpE16 ∆Mlut_03135 | This study |
| 3-M43 | trpE16 G341241A (Mlut_03135) | This study |
| 7-M89 | trpE16 C1061676T (Mlut_09880) | This study |
| 7-M93 | trpE16 C1063999T (Mlut_09900) | This study |
| 7-M28 | trpE16 G1064341A (Mlut_09900) | This study |
| 17-M33 | trpE16 C226684T (Mlut_02150) | This study |
| 13-M27 | trpE16 G1606810A (Mlut_14650) | This study |
| 13-M89 | trpE16 G1607081A (Mlut_14660) | This study |
| 13-M1090 | trpE16 G1607117A (Mlut_14660) | This study |
| 13-M93 | trpE16 C1607314T (Mlut_14660) | This study |
| 2-R33 | trpE16 G810889A (Mlut_07490) | This study |
| 13-R4 | trpE16 G1606821A (Mlut_14650) | This study |
| 8-R34 | trpE16 G1541663A (Mlut_14110) | This study |
| Cluster | SNP | Mutant/s | Mut_Type | Locus | ORF Orientation | Annotated Product |
|---|---|---|---|---|---|---|
| 1 | G103107A | M89, M106, MT | Transition | Mlut_00960 | > | Cof subfamily of IIB subfamily of haloacid dehalogenase superfamily |
| 1 | G103500A | M27, M2 | Transition | Mlut_00970 | < | Protein of unknown function DUF88 |
| 1 | G106197A | M33 | Transition | Mlut_00990 | < | Amino acid transporter |
| 3 | G341241A | M43, M47, M101, M1080, M42 | Transition | Mlut_03135 | < | N6-DNA Methylase |
| 3 | G341448A | M96 | Transition | Mlut_03135 | < | Hypothetical protein |
| 4 | C575082T | M93 | Transition | - | - | Intragenic sequence |
| 4 | G575822A | M28, M28B | Transition | Mlut_05280 | < | Hypothetical protein |
| 7 | C1061676T | M89, M106, MT | Transition | Mlut_09880 | > | ATP-dependent Clp protease proteolytic subunit ClpP |
| 7 | C1063999T | M93 | Transition | Mlut_09900 | > | ATP-dependent Clp protease ATP-binding subunit ClpX |
| 7 | G1064341A | M28, M28B | Transition | Mlut_09900 | > | ATP-dependent Clp protease ATP-binding subunit ClpX |
| 13 | G1606810A | M105, M27, M2 | Transition | Mlut_14650 | < | SAF domain-containing protein |
| 13 | G1607081A | M89, M106, MT | Transition | - | - | Intergenic sequence (Mlut_14660 promoter) |
| 13 | G1607117A | M1090 | Transition | - | - | Intergenic sequence (Mlut_14660 promoter) |
| 13 | C1607204T | M96 | Transition | Mlut_14660 | > | DNA-binding protein, excisionase family |
| 13 | C1607261T | M9, M7, M43, M47, M101, M44, M1080, M42, M6, M3 | Transition | Mlut_14660 | > | DNA-binding protein, excisionase family |
| 13 | C1607314T | M93 | Transition | Mlut_14660 | > | DNA-binding protein, excisionase family |
| 15 | C2060795T | M93 | Transition | Mlut_19080 | < | Flavodoxin |
| 15 | C2061073T | M33 | Transition | Mlut_19090 | < | Thiol-disulfide isomerase such as thioredoxin |
| 15 | T2061826C | M43 | Transition | Mlut_19090 | < | Thiol-disulfide isomerase such as thioredoxin |
| 17 | C226684T | M33 | Transition | Mlut_02150 | > | DNA-binding protein, excisionase family |
| 18 | G1878213A | M42, M1080 | Transition | Mlut_17340 | < | RNA polymerase sigma factor, sigma-70 family |
| Cluster | SNP | Mutant/s | Locus | Orientation | Annotated Product |
|---|---|---|---|---|---|
| 1 | C9672T | R42 | Mlut_00060 | > | DNA gyrase subunit A |
| 1 | G10325A | RE | Mlut_00060 | > | DNA gyrase subunit A |
| 2 | G810889A | R33 | Mlut_07490 | < | DNA segregation ATPase, FtsK/SpoIIIE family |
| 2 | G812767A | R13 | Mlut_07500 | > | Flp pilus assembly protein, ATPase CpaF |
| 2 | C812940T | R16 | Mlut_07500 | > | Flp pilus assembly protein, ATPase CpaF |
| 2 | G816694A | R34 | Mlut_07560 | > | hypothetical protein |
| 3 | C934586T | R21 | Mlut_08640 | > | glutamyl-tRNA synthetase |
| 3 | G935940A | R16 | Mlut_08660 | > | tRNA-Gln |
| 4 | G1011921A | R4 | Mlut_09340 | < | 2-oxo-acid dehydrogenase E1 component, homodimeric type |
| 4 | C1014740T | R13 | Mlut_09380 | > | 4-azaleucine resistance probable transporter AzlC |
| 5 | G1029395A | R7 | Mlut_09550 | < | peptide deformylase |
| 5 | G1031740A | R13 | Mlut_09580 | > | translocating P-type ATPase, Cd/Co/Hg/Pb/Zn-transporting |
| 6 | G1249315A | R42 | Mlut_11600 | < | phosphoserine phosphatase SerB |
| 6 | C1250564T | R42 | Mlut_11610 | > | ABC-type molybdenum transport system |
| 7 | G1460599A | RE | Mlut_13330 | > | 2-oxoglutarate dehydrogenase E2 component |
| 7 | C1462336 | R4 | Mlut_13350 | < | protein kinase family protein |
| 8 | G1541663A | R34 | Mlut_14110 | > | response regulator of citrate/malate metabolism |
| 8 | C1542830T | R41 | Mlut_14120 | < | acyl-CoA synthetase/AMP-acid ligase |
| 9 | G1814683A | R3, R30 | - | Intergenic sequence, Mlut_16600 promoter | |
| 10 | C1962653T | R4 | Mlut_18140 | > | helicase family protein with metal-binding cysteine cluster |
| 10 | C1964134T | R13 | Mlut_18140 | > | helicase family protein with metal-binding cysteine cluster |
| 11 | C2027721T | R3, R30 | Mlut_18760 | > | tRNA (guanine-N(7)-)-methyltransferase |
| 11 | C2028876T | R34 | Mlut_18770 | < | α/β hydrolase of unknown function (DUF1023) |
| 12 | G2427638A | R3, R30 | Mlut_22740 | < | hypothetical protein |
| 13 | G1606821A | R4 | Mlut_14650 | < | SAF domain-containing protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torasso Kasem, E.J.; Angelov, A.; Werner, E.; Lichev, A.; Vanderhaeghen, S.; Liebl, W. Identification of New Chromosomal Loci Involved in com Genes Expression and Natural Transformation in the Actinobacterial Model Organism Micrococcus luteus. Genes 2021, 12, 1307. https://doi.org/10.3390/genes12091307
Torasso Kasem EJ, Angelov A, Werner E, Lichev A, Vanderhaeghen S, Liebl W. Identification of New Chromosomal Loci Involved in com Genes Expression and Natural Transformation in the Actinobacterial Model Organism Micrococcus luteus. Genes. 2021; 12(9):1307. https://doi.org/10.3390/genes12091307
Chicago/Turabian StyleTorasso Kasem, Enzo Joaquin, Angel Angelov, Elisa Werner, Antoni Lichev, Sonja Vanderhaeghen, and Wolfgang Liebl. 2021. "Identification of New Chromosomal Loci Involved in com Genes Expression and Natural Transformation in the Actinobacterial Model Organism Micrococcus luteus" Genes 12, no. 9: 1307. https://doi.org/10.3390/genes12091307
APA StyleTorasso Kasem, E. J., Angelov, A., Werner, E., Lichev, A., Vanderhaeghen, S., & Liebl, W. (2021). Identification of New Chromosomal Loci Involved in com Genes Expression and Natural Transformation in the Actinobacterial Model Organism Micrococcus luteus. Genes, 12(9), 1307. https://doi.org/10.3390/genes12091307