
Pushing the Boundaries: Forensic DNA Phenotyping Challenged by Single-Cell Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Staining and Counting the Cells
2.2. Sorting the Cells
Preventing Contamination
2.3. Library Preparation and Sequencing
2.3.1. Library Amplification
2.4. Data Analysis
2.4.1. Phenotype and Ancestry Prediction
2.4.2. Interpretation Models
3. Results
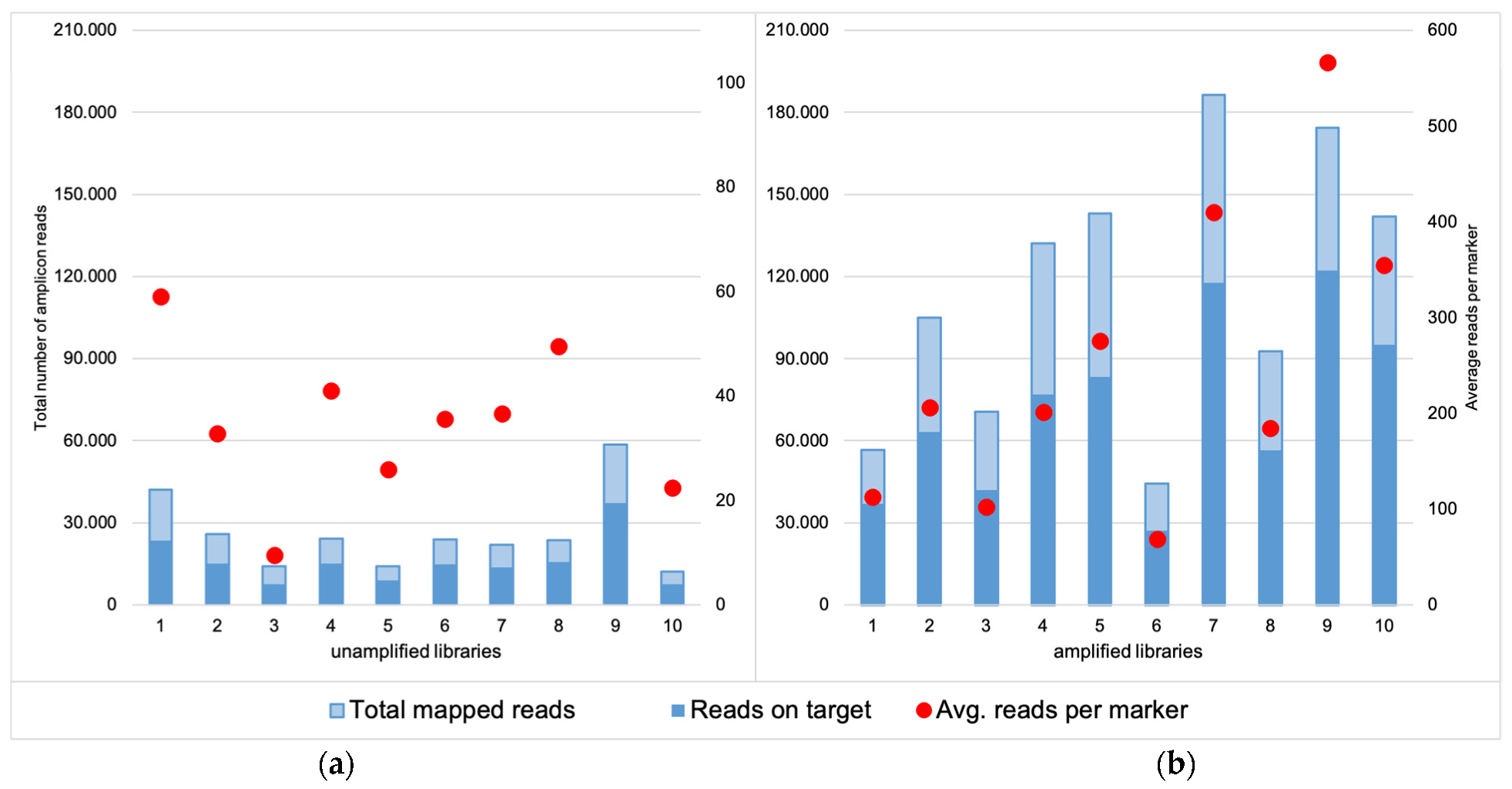
3.1. Cell Groups
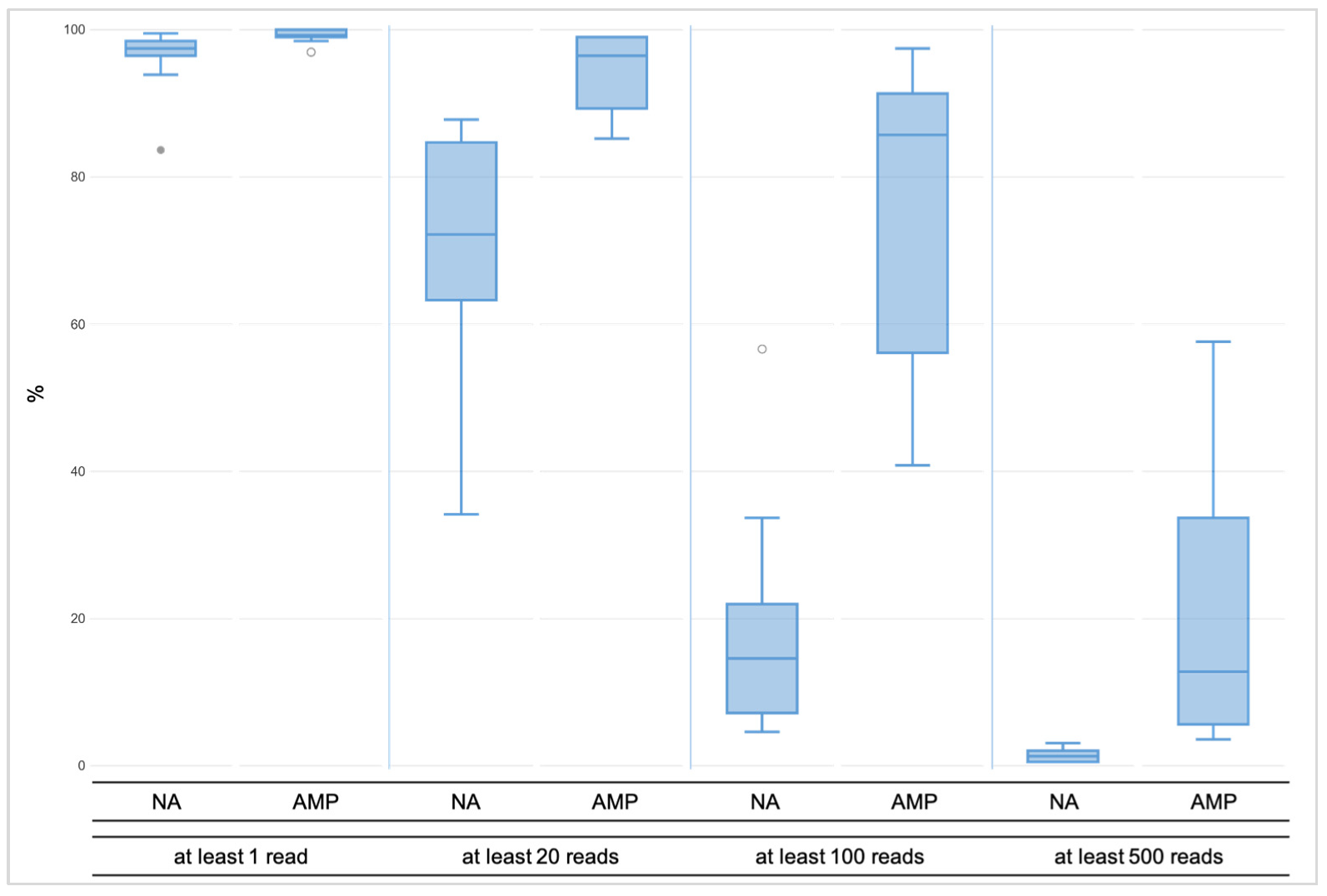
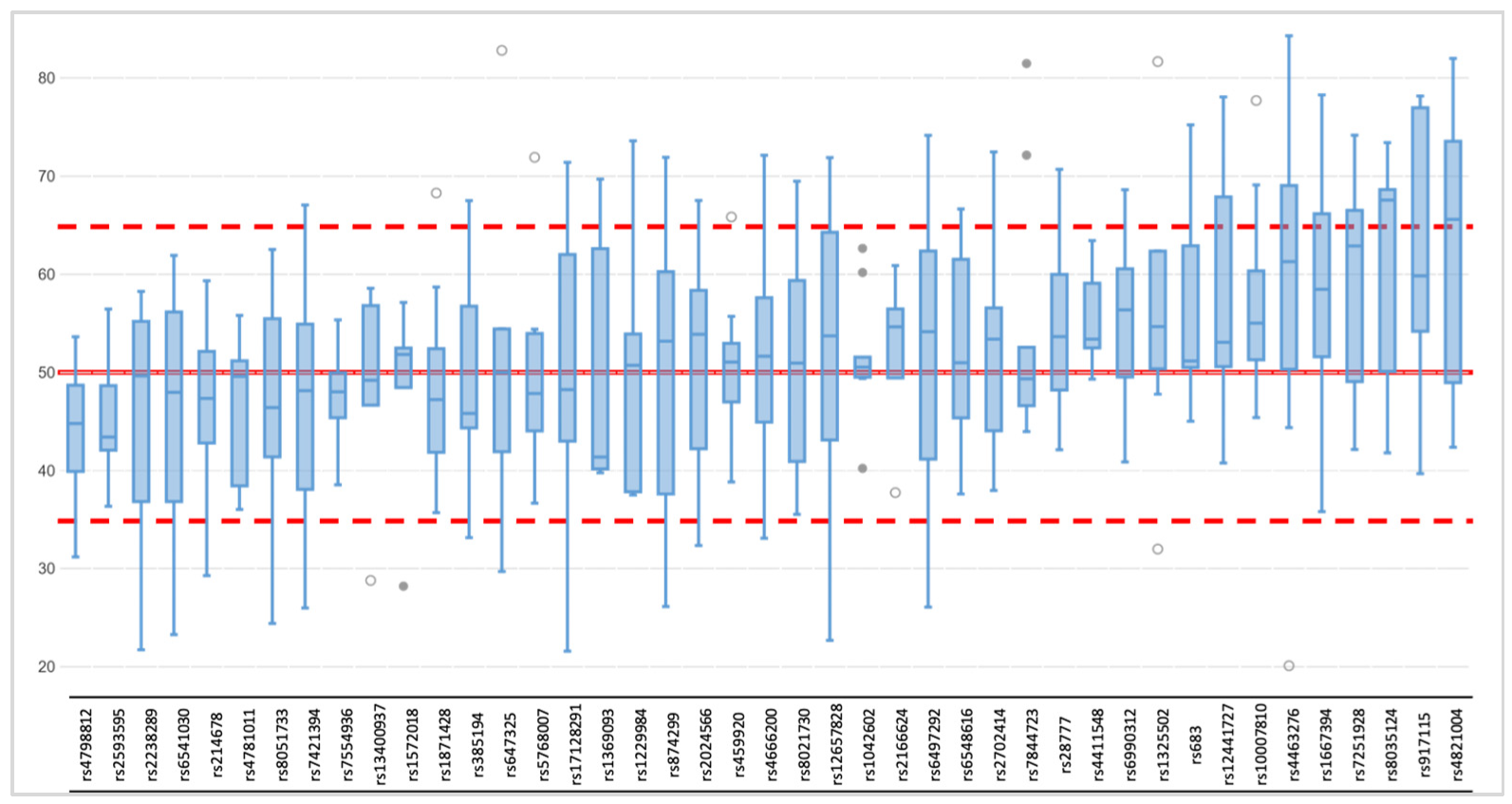
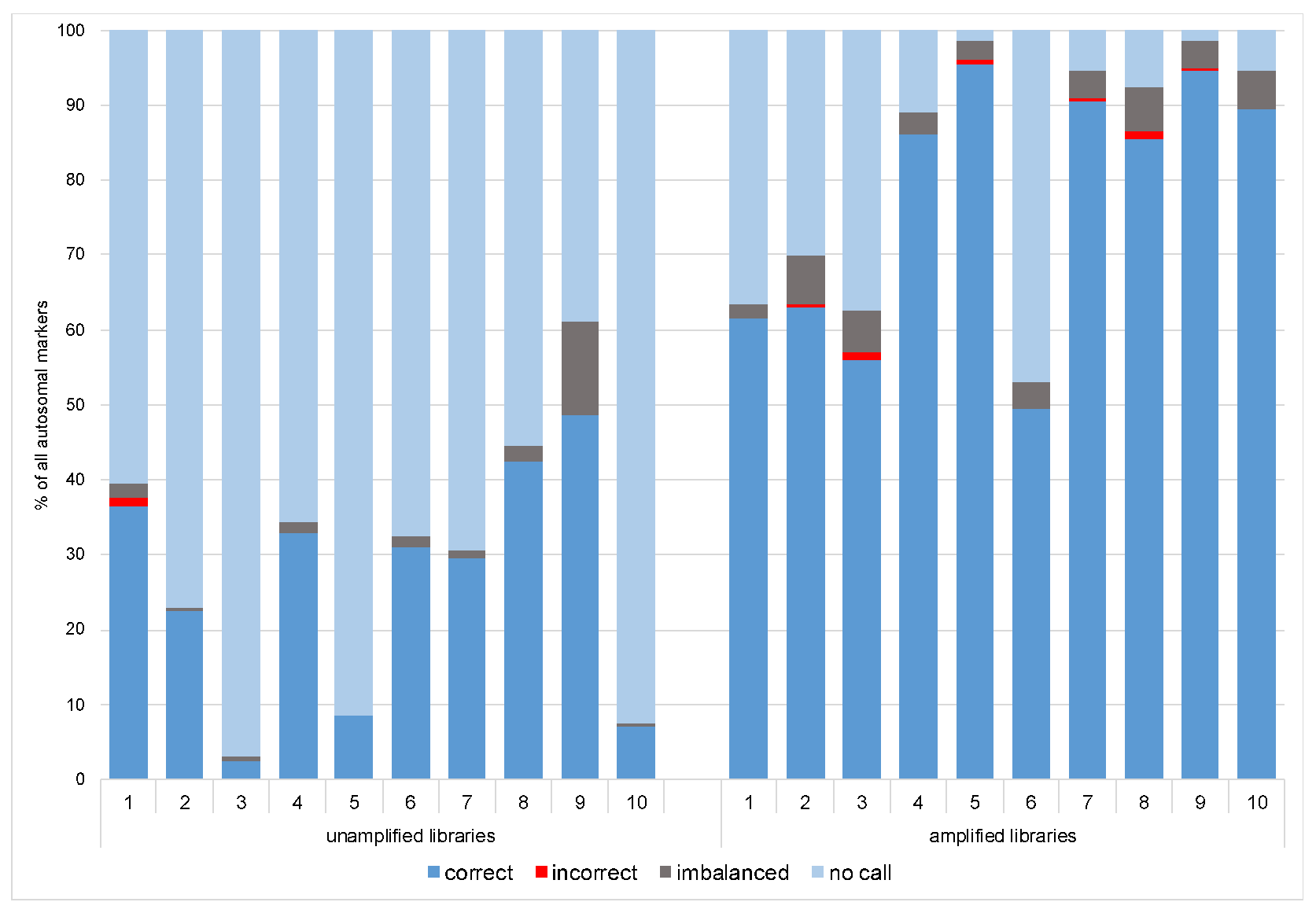
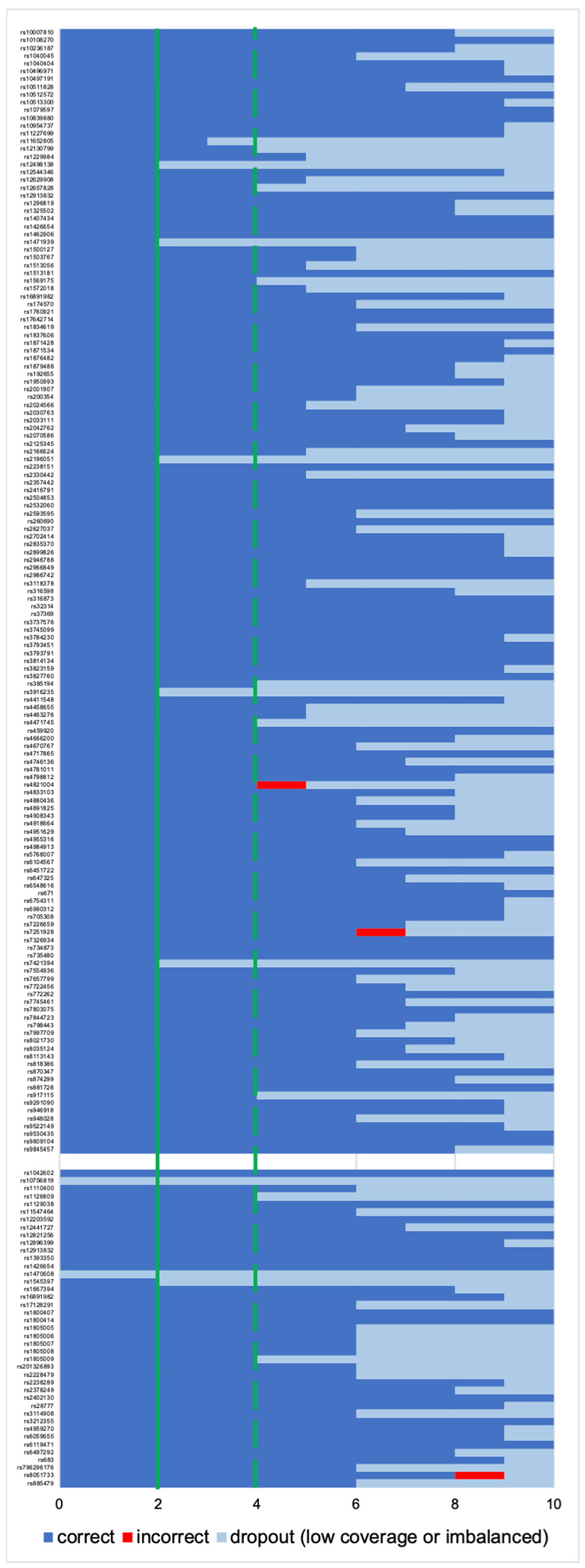
3.1.1. Coverage, Allele Frequency, and Genotype Calling
3.1.2. Phenotype and Ancestry Prediction
3.2. Single Cells
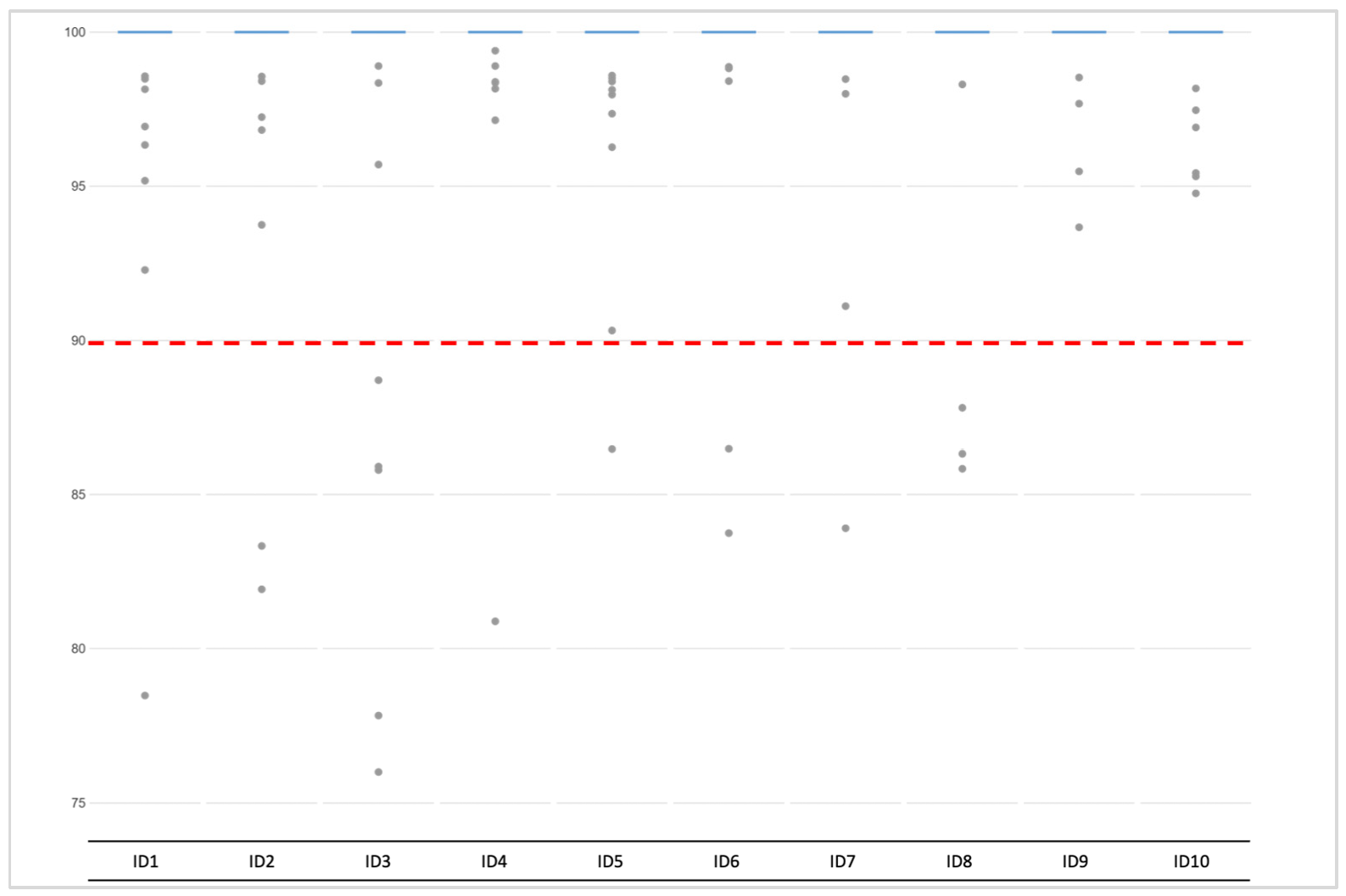
3.2.1. Coverage, allele frequency, base misincorporation rates, and genotype calling
3.2.2. Data Interpretation–“Basic” and “Conservative” Models
3.2.3. Data Interpretation-Single-Cell Based Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macaulay, I.C.; Voet, T. Single Cell Genomics: Advances and Future Perspectives. PLoS Genet. 2014, 10, e1004126. [Google Scholar] [CrossRef] [Green Version]
- Seven Technologies to Watch in 2021. Available online: https://www.nature.com/articles/d41586-021-00191-z (accessed on 26 July 2021).
- Elliott, K.; Hill, D.S.; Lambert, D.; Burroughes, T.R.; Gill, P. Use of laser microdissection greatly improves the recovery of DNA from sperm on microscope slides. Forensic Sci. Int. 2003, 137, 28–36. [Google Scholar] [CrossRef]
- Verdon, T.J.; Mitchell, R.J.; Chen, W.; Xiao, K.; van Oorschot, R.A.H. FACS separation of non-compromised forensically relevant biological mixtures. Forensic Sci. Int. Genet. 2015, 14, 194–200. [Google Scholar] [CrossRef]
- Stokes, N.A.; Stanciu, C.E.; Brocato, E.R.; Ehrhardt, C.J.; Greenspoon, S.A. Simplification of complex DNA profiles using front end cell separation and probabilistic modeling. Forensic Sci. Int. Genet. 2018, 36, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Dean, L.; Kwon, Y.J.; Philpott, M.K.; Stanciu, C.E.; Seashols-Williams, S.J.; Dawson Cruz, T.; Sturgill, J.; Ehrhardt, C.J. Separation of uncompromised whole blood mixtures for single source STR profiling using fluorescently-labeled human leukocyte antigen (HLA) probes and fluorescence activated cell sorting (FACS). Forensic Sci. Int. Genet. 2015, 17, 8–16. [Google Scholar] [CrossRef]
- Auka, N.; Valle, M.; Cox, B.D.; Wilkerson, P.D.; Dawson Cruz, T.; Reiner, J.E.; Seashols-Williams, S.J. Optical tweezers as an effective tool for spermatozoa isolation from mixed forensic samples. PLoS ONE 2019, 14, e0211810. [Google Scholar] [CrossRef]
- Horsman, K.M.; Barker, S.L.R.; Ferrance, J.P.; Forrest, K.A.; Koen, K.A.; Landers, J.P. Separation of sperm and epithelial cells in a microfabricated device: Potential application to forensic analysis of sexual assault evidence. Anal. Chem. 2005, 77, 742–749. [Google Scholar] [CrossRef]
- Zhao, X.-C.; Wang, L.; Sun, J.; Jiang, B.-W.; Zhang, E.-L.; Ye, J. Isolating Sperm from Cell Mixtures Using Magnetic Beads Coupled with an Anti-PH-20 Antibody for Forensic DNA Analysis. PLoS ONE 2016, 11, e0159401. [Google Scholar] [CrossRef] [Green Version]
- Huffman, K.; Hanson, E.; Ballantyne, J. Recovery of single source DNA profiles from mixtures by direct single cell subsampling and simplified micromanipulation. Sci. Justice 2021, 61, 13–25. [Google Scholar] [CrossRef]
- Anslinger, K.; Bayer, B.; Graw, M. Deconvolution of blood-blood mixtures using DEPArrayTM separated single cell STR profiling. Rechtsmedizin 2019, 29, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Philippron, A.; Depypere, L.; Oeyen, S.; Laere, B.D.; Vandeputte, C.; Nafteux, P.; de Preter, K.; Pattyn, P. Evaluation of a marker independent isolation method for circulating tumor cells in esophageal adenocarcinoma. PLoS ONE 2021, 16, e0251052. [Google Scholar] [CrossRef]
- Salvianti, F.; Gelmini, S.; Mancini, I.; Pazzagli, M.; Pillozzi, S.; Giommoni, E.; Brugia, M.; di Costanzo, F.; Galardi, F.; de Luca, F.; et al. Circulating tumour cells and cell-free DNA as a prognostic factor in metastatic colorectal cancer: The OMITERC prospective study. Br. J. Cancer 2021, 125, 94–100. [Google Scholar] [CrossRef]
- Rossi, T.; Gallerani, G.; Angeli, D.; Cocchi, C.; Bandini, E.; Fici, P.; Gaudio, M.; Martinelli, G.; Rocca, A.; Maltoni, R.; et al. Single-Cell NGS-Based Analysis of Copy Number Alterations Reveals New Insights in Circulating Tumor Cells Persistence in Early-Stage Breast Cancer. Cancers 2020, 12, 2490. [Google Scholar] [CrossRef]
- Boyer, M.; Cayrefourcq, L.; Garima, F.; Foulongne, V.; Dereure, O.; Alix-Panabières, C. Circulating Tumor Cell Detection and Polyomavirus Status in Merkel Cell Carcinoma. Sci. Rep. 2020, 10, 1612. [Google Scholar] [CrossRef] [Green Version]
- Meloni, V.; Lombardi, L.; Aversa, R.; Barni, F.; Berti, A. Optimization of STR amplification down to single cell after DEPArrayTM isolation. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 711–713. [Google Scholar] [CrossRef]
- Williamson, V.R.; Laris, T.M.; Romano, R.; Marciano, M.A. Enhanced DNA mixture deconvolution of sexual offense samples using the DEPArray™ system. Forensic Sci. Int. Genet. 2018, 34, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.R.L.; Myers, D.; Xavier, H.E.; Marciano, M.A. Revisiting single cell analysis in forensic science. Sci. Rep. 2021, 11, 7054. [Google Scholar] [CrossRef]
- Fontana, F.; Rapone, C.; Bregola, G.; Aversa, R.; de Meo, A.; Signorini, G.; Sergio, M.; Ferrarini, A.; Lanzellotto, R.; Medoro, G.; et al. Isolation and genetic analysis of pure cells from forensic biological mixtures: The precision of a digital approach. Forensic Sci. Int. Genet. 2017, 29, 225–241. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.M.; Prainsack, B.; Kayser, M. The use of forensic DNA phenotyping in predicting appearance and biogeographic ancestry. Dtsch. Arztebl. Int. 2019, 116, 873–880. [Google Scholar] [CrossRef]
- Eduardoff, M.; Gross, T.E.; Santos, C.; de la Puente, M.; Ballard, D.; Strobl, C.; Børsting, C.; Morling, N.; Fusco, L.; Hussing, C.; et al. Inter-laboratory evaluation of the EUROFORGEN Global ancestry-informative SNP panel by massively parallel sequencing using the Ion PGM™. Forensic Sci. Int. Genet. 2016, 23, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Breslin, K.; Wills, B.; Ralf, A.; Garcia, M.V.; Kukla-Bartoszek, M.; Pospiech, E.; Freire-Aradas, A.; Xavier, C.; Ingold, S.; De La Puente, M.; et al. HIrisPlex-S system for eye, hair, and skin color prediction from DNA: Massively parallel sequencing solutions for two common forensically used platforms. Forensic Sci. Int. Genet. 2019, 43, 102152. [Google Scholar] [CrossRef]
- Xavier, C.; De La Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.E.; Revoir, A.; Pośpiech, E.; Kartasińska, E.; et al. Development and validation of the VISAGE AmpliSeq basic tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet. 2020, 48, 102336. [Google Scholar] [CrossRef]
- Diepenbroek, M.; Bayer, B.; Schwender, K.; Schiller, R.; Lim, J.; Lagacé, R.; Anslinger, K. Evaluation of the Ion AmpliSeq™ PhenoTrivium Panel: MPS-Based Assay for Ancestry and Phenotype Predictions Challenged by Casework Samples. Genes 2020, 11, 1398. [Google Scholar] [CrossRef] [PubMed]
- Precision ID SNP Panels with the HID Ion S5™/HID Ion GeneStudio™ S5 System Application Guide. Pub. No. MAN0017767. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017767_PrecisionID_SNP_Panels_S5_UG.pdf (accessed on 1 July 2021).
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Wootton, S.; Vijaychander, S.; Hasegawa, R.; Deng, J.; Lackey, A.; Gabriel, M.; Lagacé, R.; Lim, J. Analytical Improvements in Biogeographic Ancestry Inference. Presented at the 28th Congress of the International Society for Forensic Genetics; Available online: https://www.thermofisher.com/de/de/home/products-and-services/promotions/isfg.html (accessed on 26 July 2021).
- Pereira, V.; Mogensen, H.S.; Børsting, C.; Morling, N. Evaluation of the Precision ID Ancestry Panel for crime case work: A SNP typing assay developed for typing of 165 ancestral informative markers. Forensic Sci. Int. Genet. 2017, 28, 138–145. [Google Scholar] [CrossRef]
- Liu, F.; van Duijn, K.; Vingerling, J.R.; Hofman, A.; Uitterlinden, A.G.; Cecile, A.; Janssens, J.W.; Kayser, M. Eye color and the prediction of complex phenotypes from genotypes. Curr. Biol. 2009, 19, 192–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, O.S.; Lunn, M.M.B.; Garcia, S.L.; Kjærbye, A.B.; Morling, N.; Børsting, C.; Andersen, J.D. Association between brown eye colour in rs12913832:GG individuals and SNPs in TYR, TYRP1, and SLC24A4. PLoS ONE 2020, 15, e0239131. [Google Scholar] [CrossRef]
- Meyer, O.S.; Salvo, N.M.; Kjærbye, A.; Kjersem, M.; Andersen, M.M.; Sørensen, E.; Ullum, H.; Janssen, K.; Morling, N.; Børstin, C.; et al. Prediction of Eye Colour in Scandinavians Using the EyeColour11 (EC11) SNP Set. Genes 2021, 12, 821. [Google Scholar] [CrossRef]
- Branicki, W.; Brudnik, U.; Draus-Barini, J.; Kupiec, T.; Wojas-Pelc, A. Association of the SLC45A2 gene with physiological human hair colour variation. J. Hum. Genet. 2008, 53, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.D.; Mbarek, H.; Willemsen, G.; Dolan, C.V.; Fedko, I.O.; Abdellaoui, A. Heritability and Genome-Wide Association Studies for Hair Color in a Dutch Twin Family Based Sample. Genes 2015, 13, 559–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.; Chaitanya, L.; Breslin, K.; Muralidharan, C.; Bronikowska, A.; Pospiech, E.; Koller, J.; Kovatsi, L.; Wollstein, A.; Branicki, W.; et al. Global skin colour prediction from DNA. Hum. Genet. 2017, 136, 847–863. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, L.; Breslin, K.; Zuñiga, S.; Wirken, L.; Pośpiech, E.; Kukla-Bartoszek, M.; Sijen, T.; De Knijff, P.; Liu, F.; Branicki, W.; et al. The HIrisPlex-S system for eye, hair and skin colour prediction from DNA: Introduction and forensic developmental validation. Forensic Sci. Int. Genet. 2018, 35, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Benschop, C.C.; Hoogenboom, J.; Hovers, P.; Slagter, M.; Kruise, D.; Parag, R.; Steensma, K.; Slooten, K.; Nagel, J.H.; Dieltjes, P.; et al. DNAxs/DNAStatistX: Development and validation of a software suite for the data management and probabilistic interpretation of DNA profiles. Forensic Sci. Int. Genet. 2019, 42, 81–89. [Google Scholar] [CrossRef]
- Bleka, O.; Storvik, G.; Gill, P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic Sci. Int. Genet. 2016, 21, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Bright, J.-A.; Cheng, K.; Kerr, Z.; McGovern, C.; Kelly, H.; Moretti, T.R.; Smith, M.A.; Bieber, F.R.; Budowle, B.; Coble, M.D.; et al. STRmix™ collaborative exercise on DNA mixture interpretation. Forensic Sci. Int. Genet. 2019, 40, 1–8. [Google Scholar] [CrossRef]
- Anslinger, K.; Bayer, B.; von Mariassy, D. Application of DEPArrayTM technology for the isolation of white blood cells from cell mixtures in chimerism analysis. Rechsmedizin 2018, 28, 134–137. [Google Scholar] [CrossRef]
- Balding, D.J. Evaluation of mixed-source, low-template DNA profiles in forensic science. PNAS 2013, 110, 12241–12246. [Google Scholar] [CrossRef] [Green Version]
- Balding, D.J.; Buckleton, J. Interpreting low template DNA profiles. Forensic Sci. Int. Genet. 2009, 4, 1–10. [Google Scholar] [CrossRef]
- Kanokwongnuwut, P.; Martin, B.; Taylor, D.; Kirkbride, K.P.; Linacre, A. How many cells are required for successful DNA profiling? Forensic Sci. Int. Genet. 2021, 51, 102453. [Google Scholar] [CrossRef]
- Haned, H.; Slooten, K.; Gill, P. Exploratory data analysis for the interpretation of low template DNA mixtures. Forensic Sci. Int. Genet. 2012, 6, 762–774. [Google Scholar] [CrossRef]
- Pfeifer, C.M.; Klein-Unseld, R.; Klintschar, M.; Wiegand, P. Comparison of different interpretation strategies for low template DNA mixtures. Forensic Sci. Int. Genet. 2012, 6, 716–722. [Google Scholar] [CrossRef]
- Benschop, C.C.G.; Haned, H.; de Blaeij, T.J.P.; Meulenbroek, A.J.; Sijen, T. Assessment of mock cases involving complex low template DNA mixtures: A descriptive study. Forensic Sci. Int. Genet. 2012, 6, 697–707. [Google Scholar] [CrossRef]
- Pośpiech, E.; Kukla-Bartoszek, M.; Karłowska-Pik, J.; Zieliński, P.; Woźniak, A.; Boroń, M.; Dąbrowski, M.; Zubańska, M.; Jarosz, A.; Grzybowski, T.; et al. Exploring the possibility of predicting human head hair greying from DNA using whole-exome and targeted NGS data. BMC Genom. 2020, 21, 538. [Google Scholar] [CrossRef] [PubMed]
- Kukla-Bartoszek, M.; Pośpiech, E.; Woźniak, A.; Boroń, M.; Karłowska-Pik, J.; Teisseyre, P.; Zubańska, M.; Bronikowska, A.; Grzybowski, T.; Płoski, R.; et al. DNA-based predictive models for the presence of freckles. Forensic Sci. Int. Genet. 2019, 42, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Meyer, O.S.; Andersen, J.D.; Børsting, C. Presentation of the Human Pigmentation (HuPi) AmpliSeq™ custom panel. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 478–479. [Google Scholar] [CrossRef]
- Xavier, C.; de la Puente, M.; Phillips, C.; Eduardoff, M.; Heidegger, A.; Mosquera-Miguel, A.; Freire-Aradas, A.; Lagace, R.; Wootton, S.; Power, D.; et al. Forensic evaluation of the Asia Pacific ancestry-informative MAPlex assay. Forensic Sci. Int. Genet. 2020, 48, 102344. [Google Scholar] [CrossRef]
- Jia, J.; Wei, Y.-L.; Qin, C.-J.; Hu, L.; Wan, L.-H.; Li, C.-X. Developing a novel panel of genome-wide ancestry informative markers for bio-geographical ancestry estimates. Forensic Sci. Int. Genet. 2014, 8, 187–194. [Google Scholar] [CrossRef]
- Pereira, R.; Phillips, C.; Pinto, N.; Santos, C.; Dos Santos, S.E.B.; Amorim, A.; Carracedo, A.; Gusmão, L. Straightforward Inference of Ancestry and Admixture Proportions through Ancestry-Informative Insertion Deletion Multiplexing. PLoS ONE 2012, 7, e29684. [Google Scholar] [CrossRef] [Green Version]
- Palencia-Madrid, L.; Xavier, C.; de la Puente, M.; Hohoff, C.; Phillips, C.; Kayser, M.; Parson, W. Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System. Genes 2020, 11, 708. [Google Scholar] [CrossRef]
- Eduardoff, M.; Santos, C.; de la Puente, M.; Gross, T.; Fondevila, M.; Strobl, C.; Sobrino, B.; Ballard, D.; Schneider, P.; Carracedo, A.; et al. Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the Ion PGM™. Forensic Sci. Int. Genet. 2015, 17, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralf, A.; Kayser, M. Investigative DNA analysis of two-person mixed crime scene trace in a murder case. Forensic Sci. Int. Genet. 2021, 54, 102557. [Google Scholar] [CrossRef]
- Børsting, C.; Mogensen, H.S.; Morling, N. Forensic genetic SNP typing of low-template DNA and highly degraded DNA from crime case samples. Forensic Sci. Int. Genet. 2013, 7, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Elena, S.; Alessandro, A.; Ignazio, C.; Sharon, W.; Luigi, R.; Andrea, B. Revealing the challenges of low template DNA analysis with the prototype Ion AmpliSeq™ Identity panel v2.3 on the PGM™ Sequencer. Forensic Sci. Int. Genet. 2016, 22, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Butler Gettings, K.; Kiesler, K.M.; Vallone, P.M. Performance of a next generation sequencing SNP assay on degraded DNA. Forensic Sci. Int. Genet. 2015, 19, 1–9. [Google Scholar] [CrossRef]
- Alessandrini, F.; Caucci, S.; Onofri, V.; Melchionda, F.; Tagliabracci, A.; Bagnarelli, P.; di Sante, L.; Turchi, C.; Menzo, S. Evaluation of the Ion AmpliSeq SARS-CoV-2 Research Panel by Massive Parallel Sequencing. Genes 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Siji, A.; Karthik, K.N.; Chhotusing Pardeshi, V.; Hari, P.S.; Vasudevan, A. Targeted gene panel for genetic testing of south Indian children with steroid resistant nephrotic syndrome. BMC Med. Genet. 2018, 19, 200. [Google Scholar] [CrossRef]
- Kluska, A.; Balabas, A.; Paziewska, A.; Kulecka, M.; Nowakowska, D.; Mikula, M.; Ostrowski, J. New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC Med. Genom. 2015, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Tavira, B.; Gómez, J.; Santos, F.; Gil, H.; Alvarez, V.; Coto, E. A labor- and cost-effective non-optical semiconductor (Ion Torrent) next-generation sequencing of the SLC12A3 and CLCNKA/B genes in Gitelman's syndrome patients. J. Hum. Genet. 2014, 59, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Meiklejohn, K.A.; Robertson, J.M. Evaluation of the Precision ID Identity Panel for the Ion Torrent ™ PGM ™ sequencer. Forensic Sci. Int. Genet. 2017, 31, 48–56. [Google Scholar] [CrossRef]
- Turchi, C.; Previderè, C.; Bini, C.; Carnevali, E.; Grignani, P.; Manfredi, A.; Melchionda, F.; Onofri, V.; Pelotti, S.; Robino, C.; et al. Assessment of the Precision ID Identity Panel kit on challenging forensic samples. Forensic Sci. Int. Genet. 2020, 49, 102400. [Google Scholar] [CrossRef]
- Santos, C.; Fondevila, M.; Ballard, D.; Banemann, R.; Bento, A.M.; Børsting, C.; Branicki, W.; Brisighelli, F.; Burrington, M.; Capal, T.; et al. Forensic ancestry analysis with two capillary electrophoresis ancestry informative marker (AIM) panels: Results of a collaborative EDNAP exercise. Forensic Sci. Int. Genet. 2015, 19, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, E.Y.Y.; Gahan, M.E.; McNevin, D. Prediction of biogeographical ancestry from genotype: A comparison of classifiers. Int. J. Leg. Med. 2017, 131, 901–912. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Cells–Consensus | ||||||
|---|---|---|---|---|---|---|---|
| 20 | 10 | 5 | 1 “basic” | 1 “conservative” | |||
| Eye color | blue | 0.000 | 0.000 | ||||
| inter | 0.002 | 0.002 | |||||
| brown | 0.998 | 0.998 | |||||
| Hair color and shade | blond | 0.003 | 0.003 | ||||
| brown | 0.337 | 0.337 | |||||
| red | 0.000 | 0.000 | |||||
| black | 0.661 | 0.661 | |||||
| light | 0.005 | 0.005 | |||||
| dark | 0.995 | 0.995 | |||||
| Skin color | very pale | 0.001 | 0.001 | ||||
| pale | 0.008 | 0.008 | |||||
| inter | 0.275 | 0.275 | 0.284 | ||||
| dark | 0.696 | 0.687 | 0.676 | ||||
| dark to black | 0.021 | 0.029 | 0.030 | ||||
| Admixture |  |  |  |  |  |  | |
| Ref. | Single Cells | AUC Loss | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID4 | ID5 | ID7 | ID8 | ID9 | ID10 | ID4 | ID5 | ID7 | ID8 | ID9 | ID10 | |||
| Eye color | blue | 0.000 | 0.000 | 0.003 | 0.000 | |||||||||
| inter | 0.002 | 0.012 | 0.002 | 0.012 | 0.000 | |||||||||
| brown | 0.998 | 0.988 | 0.998 | 0.003 | 0.000 | |||||||||
| Hair color and shade | blond | 0.003 | 0.014 | 0.003 | 0.006 | 0.000 | 0.004 | |||||||
| brown | 0.337 | 0.521 * | 0.337 | 0.378 | 0.337 | 0.343 | 0.007 | 0.000 | 0.003 | |||||
| red | 0.000 | 0.000 | 0.028 | 0.000 | 0.027 | |||||||||
| black | 0.661 | 0.465 * | 0.661 | 0.619 | 0.661 | 0.654 | 0.002 | 0.000 | 0.001 | 0.000 | 0.001 | |||
| light | 0.005 | 0.018 | 0.005 | 0.003 | 0.005 | 0.001 | 0.000 | |||||||
| dark | 0.995 | 0.982 | 0.995 | 0.997 | 0.995 | 0.001 | 0.000 | |||||||
| Skin color | very pale | 0.001 | 0.003 | 0.000 | 0.000 | 0.001 | 0.012 | 0.004 | 0.002 | 0.003 | 0.002 | 0.002 | ||
| pale | 0.008 | 0.030 | 0.004 | 0.005 | 0.007 | 0.009 | 0.005 | 0.007 | 0.015 | 0.004 | 0.005 | 0.004 | 0.011 | |
| inter | 0.275 | 0.418 * | 0.153 | 0.172 | 0.252 | 0.278 | 0.187 | 0.003 | 0.012 | 0.001 | 0.002 | 0.001 | 0.008 | |
| dark | 0.696 | 0.520 * | 0.700 | 0.509 * | 0.710 | 0.689 | 0.430 * | 0.002 | 0.000 | 0.001 | 0.005 | 0.001 | 0.000 | |
| dark to black | 0.021 | 0.029 | 0.143 | 0.315 * | 0.030 | 0.024 | 0.377* | 0.001 | 0.001 | 0.002 | 0.001 | 0.000 | 0.002 | |
| Admixture |  |  |  |  |  |  |  | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diepenbroek, M.; Bayer, B.; Anslinger, K. Pushing the Boundaries: Forensic DNA Phenotyping Challenged by Single-Cell Sequencing. Genes 2021, 12, 1362. https://doi.org/10.3390/genes12091362
Diepenbroek M, Bayer B, Anslinger K. Pushing the Boundaries: Forensic DNA Phenotyping Challenged by Single-Cell Sequencing. Genes. 2021; 12(9):1362. https://doi.org/10.3390/genes12091362
Chicago/Turabian StyleDiepenbroek, Marta, Birgit Bayer, and Katja Anslinger. 2021. "Pushing the Boundaries: Forensic DNA Phenotyping Challenged by Single-Cell Sequencing" Genes 12, no. 9: 1362. https://doi.org/10.3390/genes12091362
APA StyleDiepenbroek, M., Bayer, B., & Anslinger, K. (2021). Pushing the Boundaries: Forensic DNA Phenotyping Challenged by Single-Cell Sequencing. Genes, 12(9), 1362. https://doi.org/10.3390/genes12091362