
Meta-Analysis of Brain Gene Expression Data from Mouse Model Studies of Maternal Immune Activation Using Poly(I:C)




Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Selection
2.2. Analysis of Gene Expression Data
2.3. Gene Ontology
2.4. Meta-Analysis Using Gene Ranking
2.5. Enrichment of Gene-Sets for Common Genetic Variants Associated with Neurodevelopmental Phenotypes
2.6. Enrichment of Gene-Sets for De Novo Mutations
2.7. Cell Type Enrichment
3. Results
3.1. Differential Gene Expression
3.2. Meta-Analysis by Gene Ranking
3.3. Cell-Type Enrichment Analysis
3.4. Enrichment for Genes Involved in Human Phenotypes
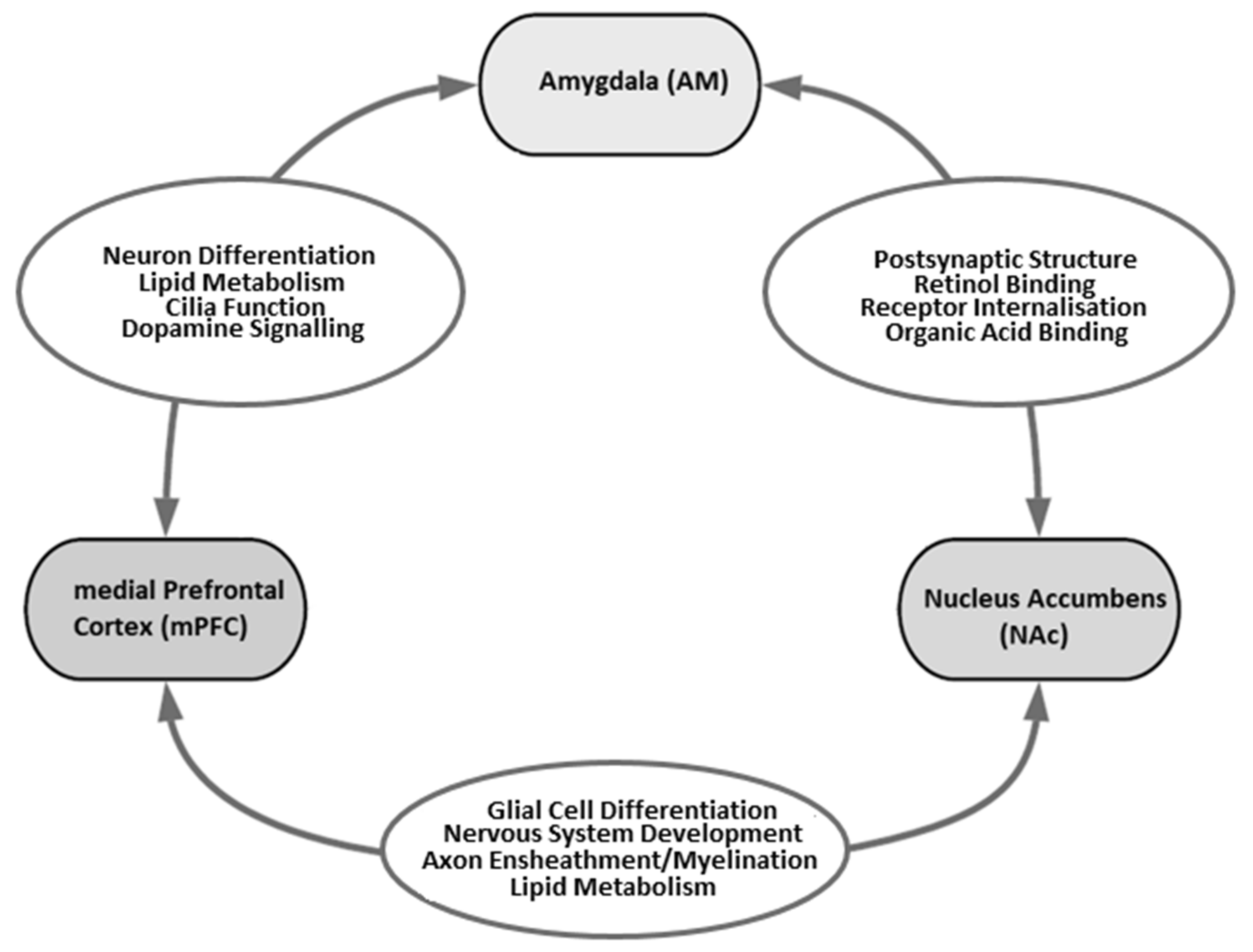
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyer, U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 2014, 75, 307–315. [Google Scholar] [CrossRef]
- Amodeo, D.A.; Lai, C.Y.; Hassan, O.; Mukamel, E.A.; Behrens, M.M.; Powell, S.B. Maternal immune activation impairs cognitive flexibility and alters transcription in frontal cortex. Neurobiol. Dis. 2019, 125, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richetto, J.; Chesters, R.; Cattaneo, A.; Labouesse, M.A.; Gutierrez, A.M.C.; Wood, T.C.; Luoni, A.; Meyer, U.; Vernon, A.; Riva, M.A. Genome-Wide Transcriptional Profiling and Structural Magnetic Resonance Imaging in the Maternal Immune Activation Model of Neurodevelopmental Disorders. Cereb. Cortex 2017, 27, 3397–3413. [Google Scholar] [CrossRef]
- Weber-Stadlbauer, U.; Richetto, J.; Labouesse, M.A.; Bohacek, J.; Mansuy, I.M.; Meyer, U. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol. Psychiatry 2017, 22, 102–112. [Google Scholar] [CrossRef]
- Sunwoo, J.S.; Jeon, D.; Lee, S.T.; Moon, J.; Yu, J.S.; Park, D.K.; Bae, J.Y.; Lee, D.Y.; Kim, S.; Jung, K.H.; et al. Maternal immune activation alters brain microRNA expression in mouse offspring. Ann. Clin. Transl. Neurol. 2018, 5, 1264–1276. [Google Scholar] [CrossRef] [Green Version]
- Mattei, D.; Ivanov, A.; Ferrai, C.; Jordan, P.; Guneykaya, D.; Buonfiglioli, A.; Schaafsma, W.; Przanowski, P.; Deuther-Conrad, W.; Brust, P.; et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl. Psychiatry 2017, 7, e1120. [Google Scholar] [CrossRef] [PubMed]
- Connor, C.M.; Dincer, A.; Straubhaar, J.; Galler, J.R.; Houston, I.B.; Akbarian, S. Maternal immune activation alters behavior in adult offspring, with subtle changes in the cortical transcriptome and epigenome. Schizophr. Res. 2012, 140, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, U.; Nyffeler, M.; Yee, B.K.; Knuesel, I.; Feldon, J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav. Immun. 2008, 22, 469–486. [Google Scholar] [CrossRef]
- Missig, G.; Robbins, J.O.; Mokler, E.L.; McCullough, K.M.; Bilbo, S.D.; McDougle, C.J.; Carlezon, W.A., Jr. Sex-dependent neurobiological features of prenatal immune activation via TLR7. Mol. Psychiatry 2020, 25, 2330–2341. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Abu-Odeh, D.; Mori, S.; Huang, H.; Oishi, K. Abnormal expression of myelination genes and alterations in white matter fractional anisotropy following prenatal viral influenza infection at E16 in mice. Schizophr. Res. 2009, 112, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, A.; Hochgerner, H.; Lonnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Haring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbett, K.A.; Hsiao, E.Y.; Kalman, S.; Patterson, P.H.; Mirnics, K. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl. Psychiatry 2012, 2, e98. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, A.; Ishima, T.; Fujita, Y.; Iwayama, Y.; Hasegawa, S.; Kawahara-Miki, R.; Maekawa, M.; Toyoshima, M.; Ushida, Y.; Suganuma, H.; et al. Dietary glucoraphanin prevents the onset of psychosis in the adult offspring after maternal immune activation. Sci. Rep. 2018, 8, 2158. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Pardinas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Karlsson Linner, R.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landen, M.; Moran, J.L.; Purcell, S.M.; Sklar, P.; Sullivan, P.F.; et al. Increased burden of ultra-rare protein-altering variants among 4877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433–1441. [Google Scholar] [CrossRef]
- Howrigan, D.P.; Rose, S.A.; Samocha, K.E.; Fromer, M.; Cerrato, F.; Chen, W.J.; Churchhouse, C.; Chambert, K.; Chandler, S.D.; Daly, M.J.; et al. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci. 2020, 23, 185–193. [Google Scholar] [CrossRef]
- Rees, E.; Han, J.; Morgan, J.; Carrera, N.; Escott-Price, V.; Pocklington, A.J.; Duffield, M.; Hall, L.S.; Legge, S.E.; Pardinas, A.F.; et al. De novo mutations identified by exome sequencing implicate rare missense variants in SLC6A1 in schizophrenia. Nat. Neurosci. 2020, 23, 179–184. [Google Scholar] [CrossRef]
- Wang, Q.; Li, M.; Yang, Z.; Hu, X.; Wu, H.M.; Ni, P.; Ren, H.; Deng, W.; Li, M.; Ma, X.; et al. Increased co-expression of genes harboring the damaging de novo mutations in Chinese schizophrenic patients during prenatal development. Sci. Rep. 2015, 5, 18209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e523. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Samocha, K.E.; Homsy, J.; Daly, M.J. Interpreting de novo Variation in Human Disease Using denovolyzeR. Curr. Protoc. Hum. Genet. 2015, 87, 7–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skene, N.G.; Grant, S.G. Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Umicevic Mirkov, M.; de Leeuw, C.A.; van den Heuvel, M.P.; Posthuma, D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 2019, 10, 3222. [Google Scholar] [CrossRef] [Green Version]
- Vehof, J.; Burger, H.; Wilffert, B.; Al Hadithy, A.; Alizadeh, B.Z.; Snieder, H. Clinical response to antipsychotic drug treatment: Association study of polymorphisms in six candidate genes. Eur. Neuropsychopharmacol. 2012, 22, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic acid signaling and neuronal differentiation. Cell Mol. Life Sci. 2015, 72, 1559–1576. [Google Scholar] [CrossRef] [Green Version]
- Jones-Villeneuve, E.M.; McBurney, M.W.; Rogers, K.A.; Kalnins, V.I. Retinoic acid induces embryonal carcinoma cells to differentiate into neurons and glial cells. J. Cell Biol. 1982, 94, 253–262. [Google Scholar] [CrossRef]
- Lin, Y.L.; Lin, Y.W.; Nhieu, J.; Zhang, X.; Wei, L.N. Sonic Hedgehog-Gli1 Signaling and Cellular Retinoic Acid Binding Protein 1 Gene Regulation in Motor Neuron Differentiation and Diseases. Int. J. Mol. Sci. 2020, 21, 4125. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, G.J.; Hu, Y.; Li, X.S.; Chen, G.Y.; Zheng, G.E.; Chen, X.; Cheng, Y. Dysregulation of Fibroblast Growth Factor 10 in the Peripheral Blood of Patients with Schizophrenia. J. Mol. Neurosci. 2019, 69, 69–74. [Google Scholar] [CrossRef]
- Bachus, S.E.; Hyde, T.M.; Herman, M.M.; Egan, M.F.; Kleinman, J.E. Abnormal cholecystokinin mRNA levels in entorhinal cortex of schizophrenics. J. Psychiatr. Res. 1997, 31, 233–256. [Google Scholar] [CrossRef]
- Sanjuan, J.; Toirac, I.; Gonzalez, J.C.; Leal, C.; Molto, M.D.; Najera, C.; De Frutos, R. A possible association between the CCK-AR gene and persistent auditory hallucinations in schizophrenia. Eur. Psychiatry 2004, 19, 349–353. [Google Scholar] [CrossRef]
- Toirac, I.; Sanjuan, J.; Aguilar, E.J.; Gonzalez, J.C.; Artigas, F.; Rivero, O.; Najera, C.; Molto, M.D.; de Frutos, R. Association between CCK-AR gene and schizophrenia with auditory hallucinations. Psychiatr. Genet. 2007, 17, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Hemmings, G.P. The CCK-A receptor gene possibly associated with auditory hallucinations in schizophrenia. Eur. Psychiatry 1999, 14, 67–70. [Google Scholar] [CrossRef]
- Zheng, C.; Fu, Q.; Shen, Y.; Xu, Q. Investigation of allelic heterogeneity of the CCK-A receptor gene in paranoid schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 741–747. [Google Scholar] [CrossRef] [PubMed]
- De Luca, V.; Tharmalingam, S.; Zai, C.; Potapova, N.; Strauss, J.; Vincent, J.; Kennedy, J.L. Association of HPA axis genes with suicidal behaviour in schizophrenia. J. Psychopharmacol. 2010, 24, 677–682. [Google Scholar] [CrossRef]
- Ribbe, K.; Ackermann, V.; Schwitulla, J.; Begemann, M.; Papiol, S.; Grube, S.; Sperling, S.; Friedrichs, H.; Jahn, O.; Sillaber, I.; et al. Prediction of the risk of comorbid alcoholism in schizophrenia by interaction of common genetic variants in the corticotropin-releasing factor system. Arch. Gen. Psychiatry 2011, 68, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.J.; Luo, T.; Zhao, Y.; Jiang, S.Z.; Xiong, J.W.; Zhan, J.Q.; Yu, B.; Yan, K.; Wei, B. Altered insulin-like growth factor-2 signaling is associated with psychopathology and cognitive deficits in patients with schizophrenia. PLoS ONE 2020, 15, e0226688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, P.; Liu, S.; Tong, D.; Cheng, M.; Wang, L.; Cheng, X. Association of polymorphisms of NAPE-PLD and FAAH genes with schizophrenia in Chinese Han population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2018, 35, 215–218. [Google Scholar] [CrossRef]
- Borglum, A.D.; Demontis, D.; Grove, J.; Pallesen, J.; Hollegaard, M.V.; Pedersen, C.B.; Hedemand, A.; Mattheisen, M.; Group Investigators; Uitterlinden, A.; et al. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol. Psychiatry 2014, 19, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, Y.; Park, J.Y.; Kim, J.E.; Kim, T.K.; Choi, J.; Lee, J.E.; Lee, E.H.; Kim, D.; Kim, K.S.; et al. Loss of Adenylyl Cyclase Type-5 in the Dorsal Striatum Produces Autistic-Like Behaviors. Mol. Neurobiol. 2017, 54, 7994–8008. [Google Scholar] [CrossRef]
- Nava, C.; Keren, B.; Mignot, C.; Rastetter, A.; Chantot-Bastaraud, S.; Faudet, A.; Fonteneau, E.; Amiet, C.; Laurent, C.; Jacquette, A.; et al. Prospective diagnostic analysis of copy number variants using SNP microarrays in individuals with autism spectrum disorders. Eur. J. Hum. Genet. 2014, 22, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Siracusano, M.; Riccioni, A.; Baratta, A.; Baldi, M.; Curatolo, P.; Mazzone, L. Autistic symptoms in Greig cephalopolysyndactyly syndrome: A family case report. J. Med. Case Rep. 2019, 13, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariminejad, A.; Yazdan, H.; Rahimian, E.; Kalhor, Z.; Fattahi, Z.; Zonooz, M.F.; Najmabadi, H.; Ashrafi, M. SZT2 mutation in a boy with intellectual disability, seizures and autistic features. Eur. J. Med. Genet. 2019, 62, 103556. [Google Scholar] [CrossRef] [PubMed]
- Hnoonual, A.; Thammachote, W.; Tim-Aroon, T.; Rojnueangnit, K.; Hansakunachai, T.; Sombuntham, T.; Roongpraiwan, R.; Worachotekamjorn, J.; Chuthapisith, J.; Fucharoen, S.; et al. Chromosomal microarray analysis in a cohort of underrepresented population identifies SERINC2 as a novel candidate gene for autism spectrum disorder. Sci. Rep. 2017, 7, 12096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhunaizi, E.; Koenekoop, R.K.; Saint-Martin, C.; Russell, L. Maternally inherited MAF variant associated with variable expression of Ayme-Gripp syndrome. Am. J. Med. Genet. A 2019, 179, 2233–2236. [Google Scholar] [CrossRef]
- Frankel, W.N.; Yang, Y.; Mahaffey, C.L.; Beyer, B.J.; O’Brien, T.P. Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 2009, 8, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Domingues, F.S.; Konig, E.; Schwienbacher, C.; Volpato, C.B.; Picard, A.; Cantaloni, C.; Mascalzoni, D.; Lackner, P.; Heimbach, A.; Hoffmann, P.; et al. Compound heterozygous SZT2 mutations in two siblings with early-onset epilepsy, intellectual disability and macrocephaly. Seizure 2019, 66, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Falcone, M.; Yariz, K.O.; Ross, D.B.; Foster, J., 2nd; Menendez, I.; Tekin, M. An amino acid deletion inSZT2 in a family with non-syndromic intellectual disability. PLoS ONE 2013, 8, e82810. [Google Scholar] [CrossRef]
- Nakamura, Y.; Togawa, Y.; Okuno, Y.; Muramatsu, H.; Nakabayashi, K.; Kuroki, Y.; Ieda, D.; Hori, I.; Negishi, Y.; Togawa, T.; et al. Biallelic mutations in SZT2 cause a discernible clinical entity with epilepsy, developmental delay, macrocephaly and a dysmorphic corpus callosum. Brain Dev. 2018, 40, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Naseer, M.I.; Alwasiyah, M.K.; Abdulkareem, A.A.; Bajammal, R.A.; Trujillo, C.; Abu-Elmagd, M.; Jafri, M.A.; Chaudhary, A.G.; Al-Qahtani, M.H. A novel homozygous mutation in SZT2 gene in Saudi family with developmental delay, macrocephaly and epilepsy. Genes Genom. 2018, 40, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhong, X.; Li, T. Novel SZT2 mutations in three patients with developmental and epileptic encephalopathies. Mol. Genet. Genom. Med. 2019, 7, e926. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, N.; Nakashima, M.; Miyauchi, A.; Yoshitomi, S.; Kimizu, T.; Ganesan, V.; Teik, K.W.; Ch’ng, G.S.; Kato, M.; Mizuguchi, T.; et al. Novel biallelic SZT2 mutations in 3 cases of early-onset epileptic encephalopathy. Clin. Genet. 2018, 93, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Tan, Y.; Zhang, X.; Wang, X.; Krystal, J.; Tabakoff, B.; Zhong, C.; Luo, X. A New Genomewide Association Meta-Analysis of Alcohol Dependence. Alcohol. Clin. Exp. Res. 2015, 39, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Wang, K.S.; Zhang, X.Y.; Li, C.S.; Zhang, F.; Wang, X.; Chen, W.; Gao, G.; Zhang, H.; Krystal, J.H.; et al. Rare SERINC2 variants are specific for alcohol dependence in individuals of European descent. Pharm. Genom. 2013, 23, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Padula, A.E.; Griffin, W.C., 3rd; Lopez, M.F.; Nimitvilai, S.; Cannady, R.; McGuier, N.S.; Chesler, E.J.; Miles, M.F.; Williams, R.W.; Randall, P.K.; et al. KCNN Genes that Encode Small-Conductance Ca2+-Activated K+ Channels Influence Alcohol and Drug Addiction. Neuropsychopharmacology 2015, 40, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Haworth, K.; Smith, F.; Zoupa, M.; Seppala, M.; Sharpe, P.T.; Cobourne, M.T. Expression of the Scube3 epidermal growth factor-related gene during early embryonic development in the mouse. Gene Expr. Patterns 2007, 7, 630–634. [Google Scholar] [CrossRef]
- Iacopetti, P.; Barsacchi, G.; Tirone, F.; Cremisi, F. Expression of the PC4 gene in the developing rat nervous system. Brain Res. 1996, 707, 293–297. [Google Scholar] [CrossRef]
- Pirity, M.K.; Locker, J.; Schreiber-Agus, N. Rybp/DEDAF is required for early postimplantation and for central nervous system development. Mol. Cell Biol. 2005, 25, 7193–7202. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.; Szabo, V.; Pirity, M.K. Absence of Rybp Compromises Neural Differentiation of Embryonic Stem Cells. Stem Cells Int. 2016, 2016, 4034620. [Google Scholar] [CrossRef] [Green Version]
- Nordstrom, T.; Andersson, L.C.; Akerman, K.E.O. Regulation of intracellular pH by electrogenic Na+/HCO3- co-transporters in embryonic neural stem cell-derived radial glia-like cells. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1037–1048. [Google Scholar] [CrossRef]
- Sahara, S.; O’Leary, D.D. Fgf10 regulates transition period of cortical stem cell differentiation to radial glia controlling generation of neurons and basal progenitors. Neuron 2009, 63, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xiao, Z.; Zheng, J.; Wu, J.; Hu, X.L.; Yang, X.; Shen, Q. ZEB1 Represses Neural Differentiation and Cooperates with CTBP2 to Dynamically Regulate Cell Migration during Neocortex Development. Cell Rep. 2019, 27, 2335–2353.e2336. [Google Scholar] [CrossRef] [Green Version]
- Garza-Manero, S.; Sindi, A.A.A.; Mohan, G.; Rehbini, O.; Jeantet, V.H.M.; Bailo, M.; Latif, F.A.; West, M.P.; Gurden, R.; Finlayson, L.; et al. Maintenance of active chromatin states by HMGN2 is required for stem cell identity in a pluripotent stem cell model. Epigenetics Chromatin 2019, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- LaMantia, A.S. Forebrain induction, retinoic acid, and vulnerability to schizophrenia: Insights from molecular and genetic analysis in developing mice. Biol. Psychiatry 1999, 46, 19–30. [Google Scholar] [CrossRef]
- Dong, L.; Li, R.; Li, D.; Wang, B.; Lu, Y.; Li, P.; Yu, F.; Jin, Y.; Ni, X.; Wu, Y.; et al. FGF10 Enhances Peripheral Nerve Regeneration via the Preactivation of the PI3K/Akt Signaling-Mediated Antioxidant Response. Front. Pharmacol. 2019, 10, 1224. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Lynn Miskus, M.; Lu, H.C. FGF-FGFR Mediates the Activity-Dependent Dendritogenesis of Layer IV Neurons during Barrel Formation. J. Neurosci. 2017, 37, 12094–12105. [Google Scholar] [CrossRef]
- Moore, A.R.; Ghiretti, A.E.; Paradis, S. A loss-of-function analysis reveals that endogenous Rem2 promotes functional glutamatergic synapse formation and restricts dendritic complexity. PLoS ONE 2013, 8, e74751. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, J.L.; Ran, Y.C.; Zhang, Y.; Yang, L.; Lin, Y.N. Expression of RGMb in brain tissue of MCAO rats and its relationship with axonal regeneration. J. Neurol. Sci. 2017, 383, 79–86. [Google Scholar] [CrossRef]
- Park, H.J.; Gonzalez-Islas, C.E.; Kang, Y.; Li, J.M.; Choi, I. Deletion of the Na/HCO3 Transporter NBCn1 Protects Hippocampal Neurons from NMDA-induced Seizures and Neurotoxicity in Mice. Sci. Rep. 2019, 9, 15981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woitecki, A.M.; Muller, J.A.; van Loo, K.M.; Sowade, R.F.; Becker, A.J.; Schoch, S. Identification of Synaptotagmin 10 as Effector of NPAS4-Mediated Protection from Excitotoxic Neurodegeneration. J. Neurosci. 2016, 36, 2561–2570. [Google Scholar] [CrossRef] [Green Version]
- Berdenis van Berlekom, A.; Muflihah, C.H.; Snijders, G.; MacGillavry, H.D.; Middeldorp, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophr Bull. 2020, 46, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium; Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.; Farh, K.H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Milanova, V.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickens, A.M.; Sen, P.; Kempton, M.J.; Barrantes-Vidal, N.; Iyegbe, C.; Nordentoft, M.; Pollak, T.; Riecher-Rossler, A.; Ruhrmann, S.; Sachs, G.; et al. Dysregulated Lipid Metabolism Precedes Onset of Psychosis. Biol. Psychiatry 2021, 89, 288–297. [Google Scholar] [CrossRef]
- Adriano, F.; Caltagirone, C.; Spalletta, G. Hippocampal volume reduction in first-episode and chronic schizophrenia: A review and meta-analysis. Neuroscientist 2012, 18, 180–200. [Google Scholar] [CrossRef]
- Baiano, M.; Perlini, C.; Rambaldelli, G.; Cerini, R.; Dusi, N.; Bellani, M.; Spezzapria, G.; Versace, A.; Balestrieri, M.; Mucelli, R.P.; et al. Decreased entorhinal cortex volumes in schizophrenia. Schizophr. Res. 2008, 102, 171–180. [Google Scholar] [CrossRef]
- Konradi, C.; Yang, C.K.; Zimmerman, E.I.; Lohmann, K.M.; Gresch, P.; Pantazopoulos, H.; Berretta, S.; Heckers, S. Hippocampal interneurons are abnormal in schizophrenia. Schizophr. Res. 2011, 131, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Rosso, P.; Iannitelli, A.; Pacitti, F.; Quartini, A.; Fico, E.; Fiore, M.; Greco, A.; Ralli, M.; Tirassa, P. Vagus nerve stimulation and Neurotrophins: A biological psychiatric perspective. Neurosci. Biobehav. Rev. 2020, 113, 338–353. [Google Scholar] [CrossRef]
- Perez, S.M.; Carreno, F.R.; Frazer, A.; Lodge, D.J. Vagal nerve stimulation reverses aberrant dopamine system function in the methylazoxymethanol acetate rodent model of schizophrenia. J. Neurosci. 2014, 34, 9261–9267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, A.; Wolff-Menzler, C.; Pfeiffer, S.; Falkai, P.; Weidinger, E.; Jobst, A.; Hoell, I.; Malchow, B.; Yeganeh-Doost, P.; Strube, W.; et al. Transcutaneous noninvasive vagus nerve stimulation (tVNS) in the treatment of schizophrenia: A bicentric randomized controlled pilot study. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 589–600. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, R.A.; Abi-Dargham, A.; Howes, O.D. Schizophrenia, Dopamine and the Striatum: From Biology to Symptoms. Trends Neurosci. 2019, 42, 205–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
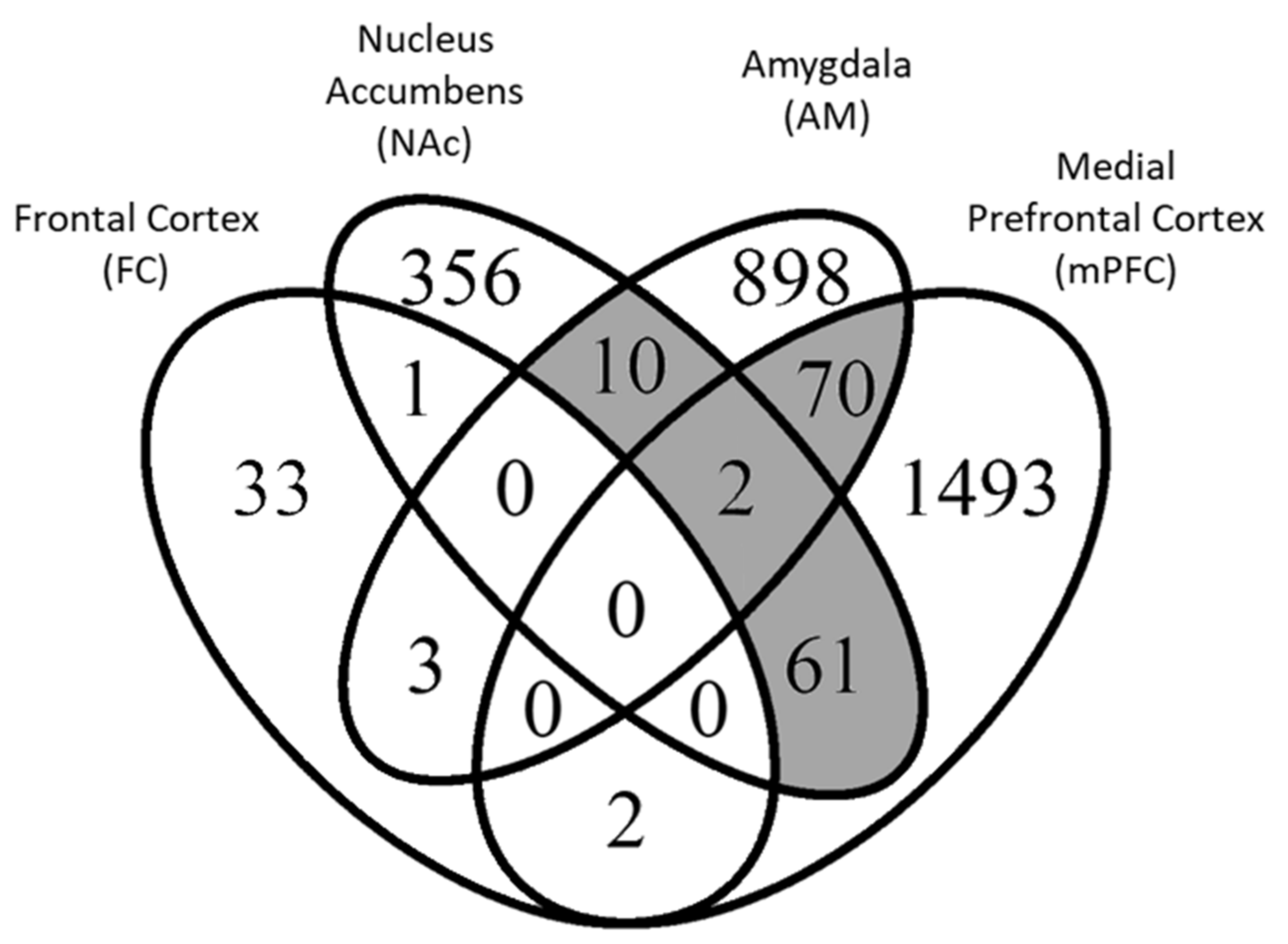

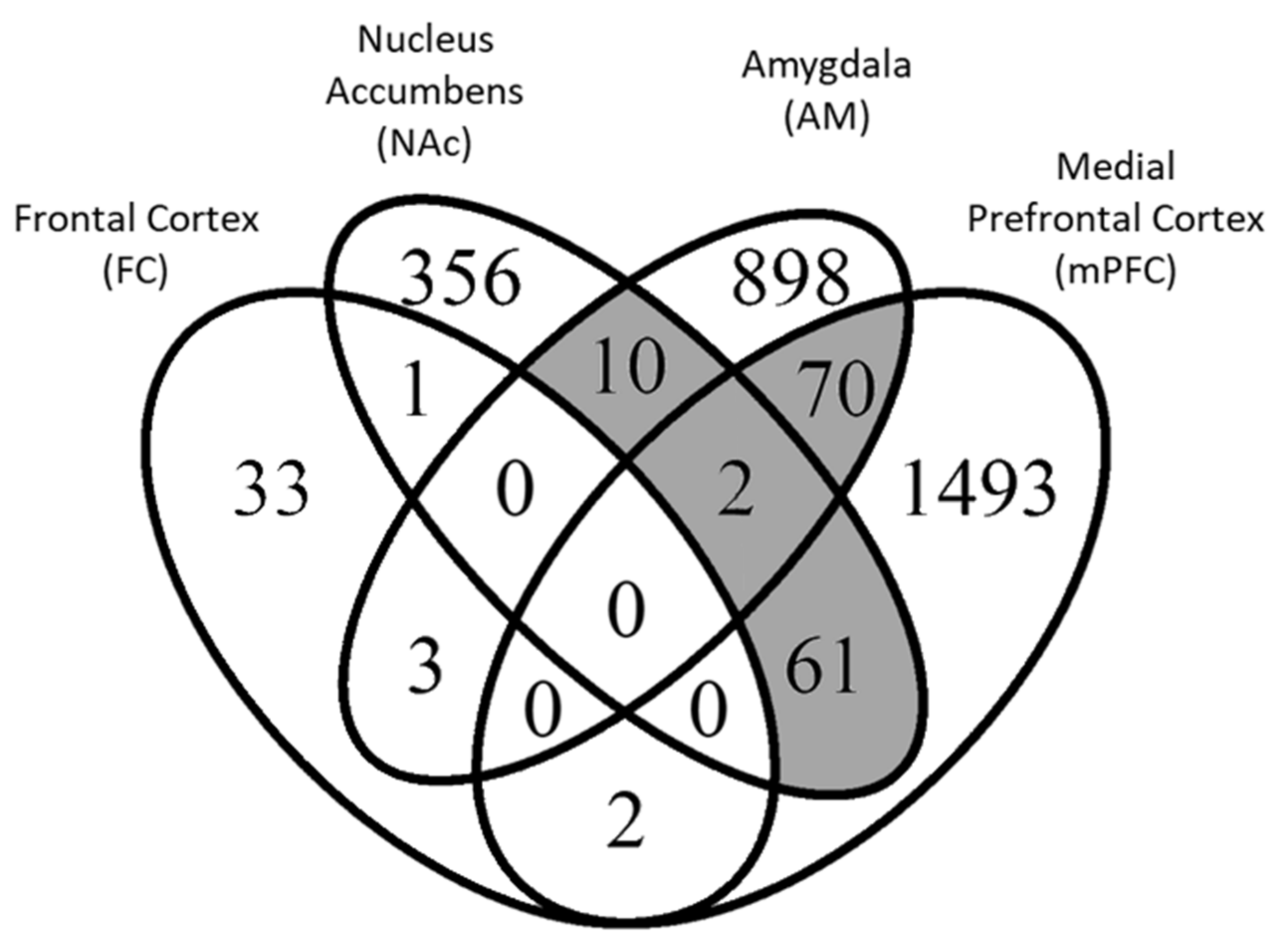{kind=link}
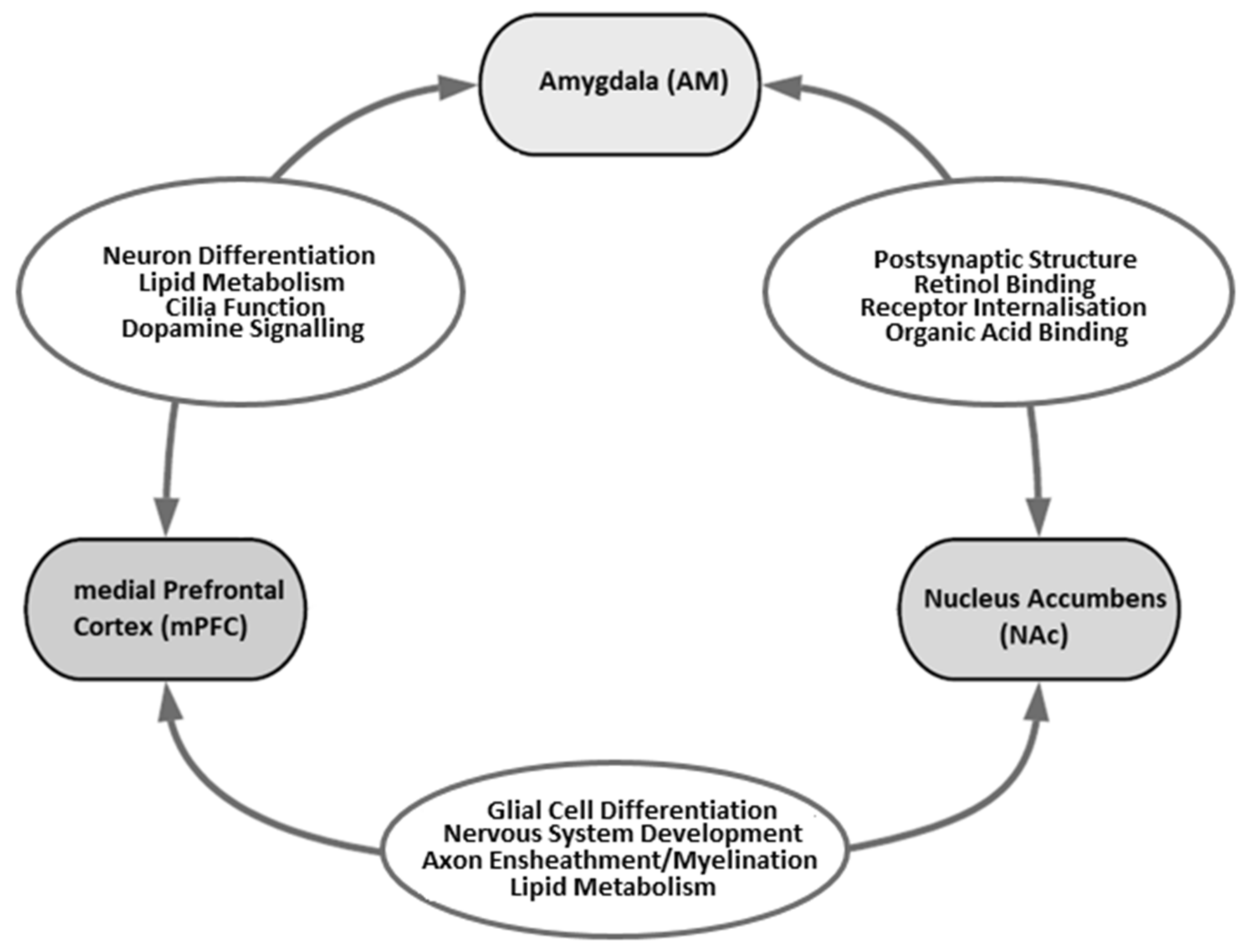{kind=link}
| Study Reference | Technique | Brain Region | # Controls/Treatments | Poly(I:C) Dose; Day of Admin. * | # Up-Regulated DEGS (FDR < 0.1) | # Down-Regulated DEGs (FDR < 0.1) |
|---|---|---|---|---|---|---|
| [2] | RNA-seq | Frontal Cortex (FC) | 8/9 | 20 mg/kg; E12.5 | 21 | 17 |
| [3] | Microarray | Prefrontal Cortex (mPFC) | 6/6 | 5 mg/kg; GD17 | 1042 | 586 |
| [3] | Microarray | Nucleus Accumbens (Nac) | 6/6 | 5 mg/kg; GD17 | 225 | 206 |
| [4] | RNA-seq | Amygdala (AM) | 6/5 | 5 mg/kg; GD9 | 521 | 462 |
| Position | Product Rank (Down) | Product Sum (Down) | Product Rank (Up) | Product Sum (Up) |
|---|---|---|---|---|
| 1 | Rybp | Scube3 | Szt2 | Cck |
| 2 | Tdrp | Hs6st2 | Fgf10 | Mical2 |
| 3 | Dach1 | Dach1 | Ttc13 | Mroh1 |
| 4 | Scube3 | Syt10 | Slc9b2 | Slc4a7 |
| 5 | Hs6st2 | Rybp | Serinc2 | Ifrd1 |
| 6 | Itga5 | Hmgn2 | Crhbp | Dysf |
| 7 | Tceal5 | Ankef1 | Rgmb | Ociad2 |
| 8 | Syt10 | Th | Maf | Fam126b |
| 9 | Adcy5 | Ogfr | Ints10 | Elmo2 |
| 10 | Dock10 | Mid1 | Igfbp7 | Hapln4 |
| 11 | Hmgn2 | Dock10 | Letmd1 | Anxa11 |
| 12 | Zeb1 | B230118H07Rik | Napepld | Rnaseh2a |
| 13 | Ass1 | Gli3 | Aifm3 | Arl5a |
| 14 | Myl12b | Aim2 | Tmem25 | Serpinb8 |
| 15 | Gli3 | Ass1 | Rnaseh2a | Slc17a5 |
| 16 | Serinc5 | Ecscr | Mospd1 | Siah3 |
| 17 | BC005624 | Bex4 | Cblb | Kcnn1 |
| 18 | Bex4 | Pbx3 | Tnpo1 | Zfp697 |
| 19 | Rem2 | Itga5 | Mroh1 | Maf |
| 20 | Smndc1 | Chmp6 | Ifrd1 | Mettl1 |
| Gene-Set | Enriched Cell Types in Zeisel Data (n = 265 Cell Types) | Enriched Cell Types in Saunders Data (n = 565 Cell Types) | |
|---|---|---|---|
| Individual Study DEGs | mPFC | -Oligodendrocytes -Excitatory Neurons (Hindbrain) -Inhibitory D2 Medium Spiny Neurons (Striatum) | -Oligodendrocytes |
| NAc | -Oligodendrocytes | -Oligodendrocytes | |
| AM | -Inhibitory D1 & D2 Medium Spiny Neurons (Striatum) -Ependymal Cells -Choroid Plexus Cells -Hypendymal Cells -Vascular Leptomeningeal Cells | -Inhibitory direct/indirect Spiny Projection Neurons (Striatum) -Ependymal Cells -Choroid Plexus Cells -Endothelial Cells | |
| Overlap DEGs | mPFC + NAc | -Oligodendrocytes | -Oligodendrocytes |
| mPFC + AM | -Inhibitory D2 Medium Spiny Neurons (Striatum) -Ependymal Cells | -Inhibitory direct/indirect Spiny Projection Neurons (Striatum) | |
| Meta-analysis DEGs | Up-regulated | -Excitatory Neurons, Pyramidal Cells (Cerebral Cortex) -Inhibitory Interneurons (Hippocampus) -Inhibitory Interneurons (Hypothalamus) | -Excitatory Neurons, Deep-layer Pyramidal cells (Frontal Cortex) -Excitatory Neurons (Posterior Cortex) -Excitatory Neurons, CA1 Principal Cells (Hippocampus) -Excitatory Neurons, Entorhinal Cortex Cells (Hippocampus) -Inhibitory Interneurons (Hippocampus) |
| Down-regulated | -Microglia | -Endothelial Stalk Cells | |
| Gene-Set | Significant Cell Types from Zeisel That Are Common between Mouse MIA and Human GWAS | Significant Cell Types from Saunders That Are Common between Mouse MIA and Human GWAS | |
|---|---|---|---|
| Individual Study DEGs | mPFC | -Excitatory Neurons (Hindbrain) [SCZ, ASD, EA] | No cell types |
| AM | -Inhibitory D1 & D2 Medium Spiny Neurons (Striatum) [EA] | -Inhibitory direct/indirect Spiny Projection Neurons (Striatum) [SCZ, EA, IQ] | |
| Meta-analysisDEGs | Upregulated | -Excitatory Neurons, Pyramidal Cells (Cerebral Cortex) [SCZ, IQ] | -Excitatory Neurons, Deep-layer Pyramidal cells (Frontal Cortex) [SCZ, IQ] -Excitatory Neurons (Posterior Cortex) [SCZ, IQ] -Excitatory Neurons, Entorhinal Cortex Cells (Hippocampus) [SCZ, IQ] -Excitatory Neurons, CA1 Principal Cells (Hippocampus) [IQ] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laighneach, A.; Desbonnet, L.; Kelly, J.P.; Donohoe, G.; Morris, D.W. Meta-Analysis of Brain Gene Expression Data from Mouse Model Studies of Maternal Immune Activation Using Poly(I:C). Genes 2021, 12, 1363. https://doi.org/10.3390/genes12091363
Laighneach A, Desbonnet L, Kelly JP, Donohoe G, Morris DW. Meta-Analysis of Brain Gene Expression Data from Mouse Model Studies of Maternal Immune Activation Using Poly(I:C). Genes. 2021; 12(9):1363. https://doi.org/10.3390/genes12091363
Chicago/Turabian StyleLaighneach, Aodán, Lieve Desbonnet, John P. Kelly, Gary Donohoe, and Derek W. Morris. 2021. "Meta-Analysis of Brain Gene Expression Data from Mouse Model Studies of Maternal Immune Activation Using Poly(I:C)" Genes 12, no. 9: 1363. https://doi.org/10.3390/genes12091363
APA StyleLaighneach, A., Desbonnet, L., Kelly, J. P., Donohoe, G., & Morris, D. W. (2021). Meta-Analysis of Brain Gene Expression Data from Mouse Model Studies of Maternal Immune Activation Using Poly(I:C). Genes, 12(9), 1363. https://doi.org/10.3390/genes12091363