
The Sound of Silence: How Silenced Chromatin Orchestrates the Repair of Double-Strand Breaks

{kind=link}
{kind=link}
{kind=link}
Abstract
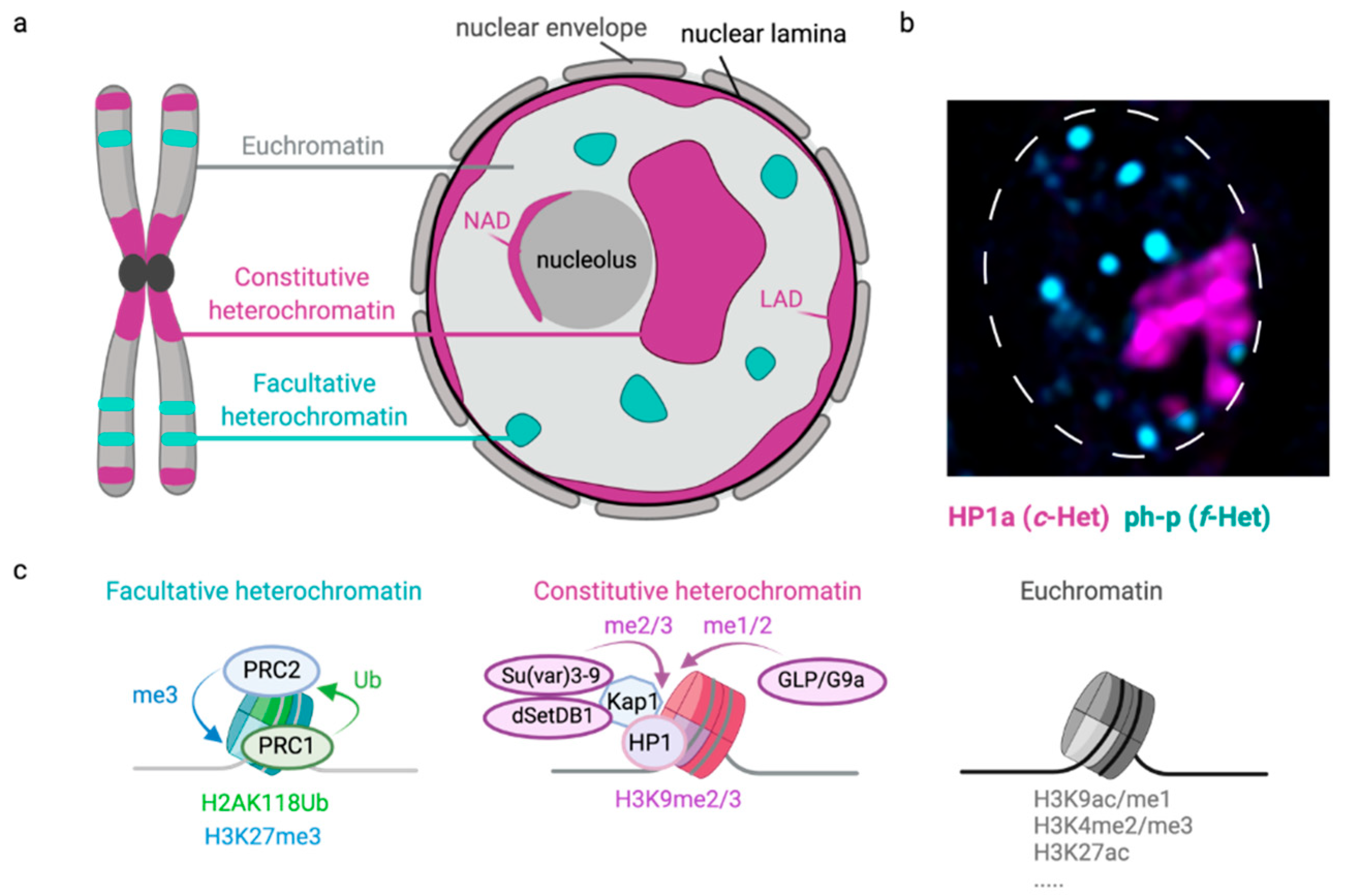
:1. Introduction
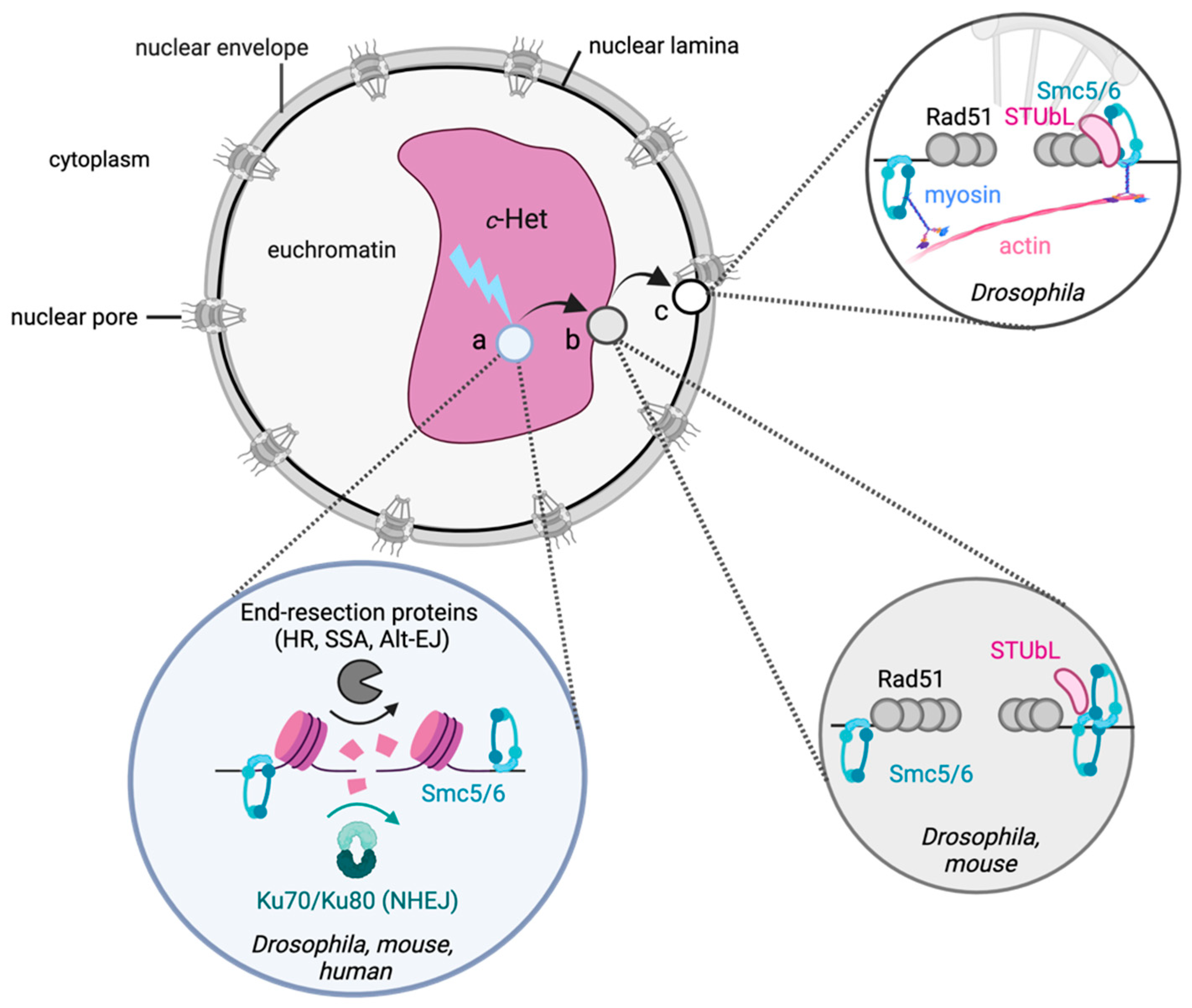
2. Constitutive Heterochromatin
2.1. General Principles of DSB Repair in c-Het
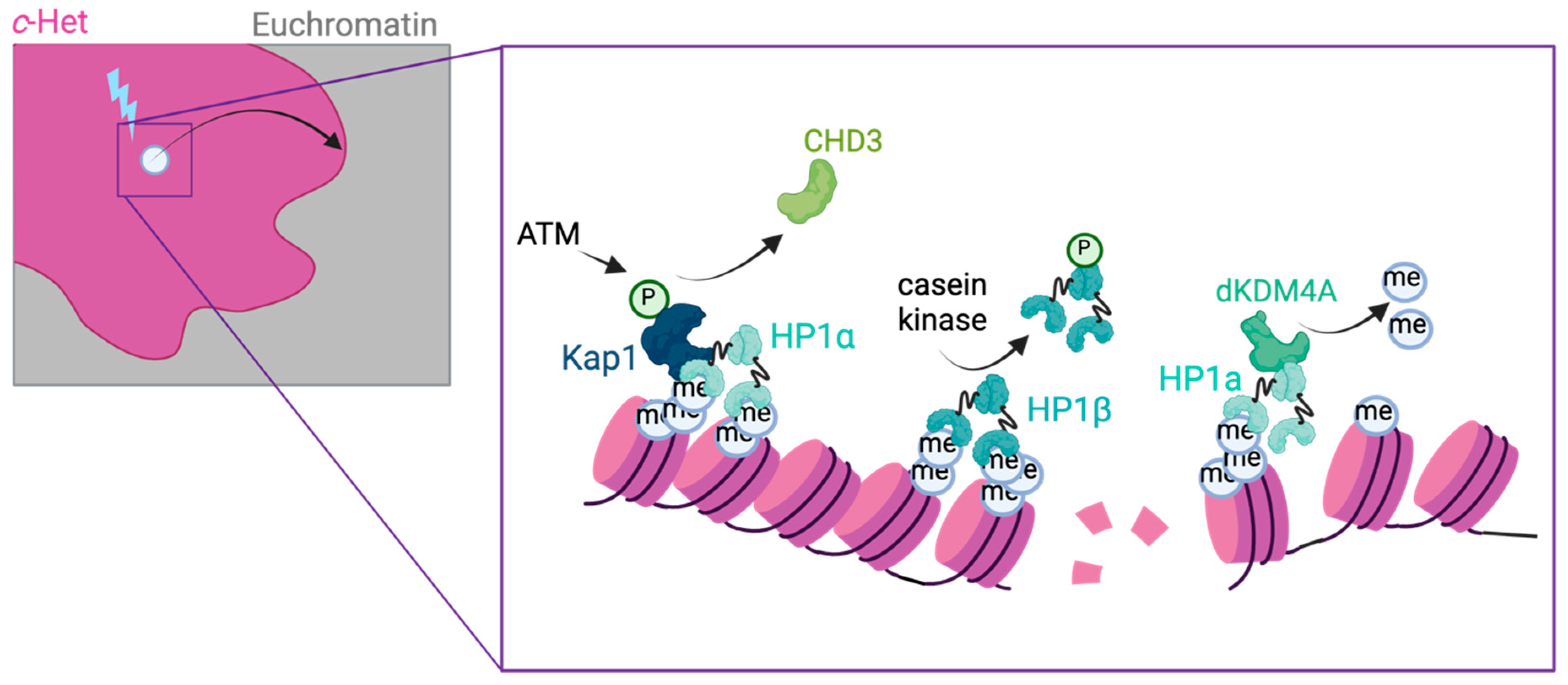
2.2. The Role of Canonical Heterochromatin Proteins in c-Het Repair
2.3. Local Chromatin Changes at c-Het DSBs
2.4. SUMOylation and the Nuclear Periphery in c-Het Repair
3. Facultative Heterochromatin
DSB Repair in f-Het
4. Lamina-Associated Domains
DSB Repair in LADs
5. Summary and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [Green Version]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, N.A.; Rass, E.; Scully, R. Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement. Trends Cancer 2015, 1, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, D.A.; Nussenzweig, A. Mechanisms driving chromosomal translocations: Lost in time and space. Oncogene 2021, 40, 4263–4270. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Mattiroli, F.; Penengo, L. Histone Ubiquitination: An Integrative Signaling Platform in Genome Stability. Trends Genet. 2021, 37, 566–581. [Google Scholar] [CrossRef]
- Clouaire, T.; Legube, G. A Snapshot on the Cis Chromatin Response to DNA Double-Strand Breaks. Trends Genet. 2019, 35, 330–345. [Google Scholar] [CrossRef] [Green Version]
- Saredi, G.; Huang, H.; Hammond, C.M.; Alabert, C.; Bekker-Jensen, S.; Forne, I.; Reveron-Gomez, N.; Foster, B.M.; Mlejnkova, L.; Bartke, T.; et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 2016, 534, 714–718. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.W.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative analysis of metazoan chromatin organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Caron, P.; Pankotai, T.; Wiegant, W.W.; Tollenaere, M.A.X.; Furst, A.; Bonhomme, C.; Helfricht, A.; de Groot, A.; Pastink, A.; Vertegaal, A.C.O.; et al. WWP2 ubiquitylates RNA polymerase II for DNA-PK-dependent transcription arrest and repair at DNA breaks. Genes Dev. 2019, 33, 684–704. [Google Scholar] [CrossRef]
- Gong, F.; Clouaire, T.; Aguirrebengoa, M.; Legube, G.; Miller, K.M. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell Biol. 2017, 216, 1959–1974. [Google Scholar] [CrossRef] [Green Version]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015, 17, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.; Breuer, G.A.; Brinkman, E.K.; van der Meulen, A.I.; Borden, S.V.; van Steensel, B.; Bindra, R.S.; LaRocque, J.R.; Karpen, G.H. A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev. 2016, 30, 1645–1657. [Google Scholar] [CrossRef] [Green Version]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.O.; Zink, D.; Durante, M.; Lobrich, M.; Taucher-Scholz, G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef]
- Plys, A.J.; Davis, C.P.; Kim, J.; Rizki, G.; Keenen, M.M.; Marr, S.K.; Kingston, R.E. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019, 33, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef]
- Strom, A.R.; Brangwynne, C.P. The liquid nucleome—Phase transitions in the nucleus at a glance. J. Cell Sci. 2019, 132, jcs235093. [Google Scholar] [CrossRef] [Green Version]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatavosian, R.; Kent, S.; Brown, K.; Yao, T.; Duc, H.N.; Huynh, T.N.; Zhen, C.Y.; Ma, B.; Wang, H.; Ren, X. Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019, 294, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Padeken, J.; Zeller, P.; Gasser, S.M. Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 2015, 31, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Heitz, E. Das Heterochromatin der Moose; Bornträger: Stuttgart, Germany, 1928. [Google Scholar]
- Passarge, E. Emil Heitz and the concept of heterochromatin: Longitudinal chromosome differentiation was recognized fifty years ago. Am. J. Hum. Genet. 1979, 31, 106–115. [Google Scholar]
- Hoskins, R.A.; Carlson, J.W.; Kennedy, C.; Acevedo, D.; Evans-Holm, M.; Frise, E.; Wan, K.H.; Park, S.; Mendez-Lago, M.; Rossi, F.; et al. Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science 2007, 316, 1625–1628. [Google Scholar] [CrossRef] [Green Version]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229–244. [Google Scholar] [CrossRef]
- Schotta, G.; Ebert, A.; Krauss, V.; Fischer, A.; Hoffmann, J.; Rea, S.; Jenuwein, T.; Dorn, R.; Reuter, G. Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 2002, 21, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Brower-Toland, B.; Riddle, N.C.; Jiang, H.; Huisinga, K.L.; Elgin, S.C. Multiple SET methyltransferases are required to maintain normal heterochromatin domains in the genome of Drosophila melanogaster. Genetics 2009, 181, 1303–1319. [Google Scholar] [CrossRef] [Green Version]
- James, T.C.; Elgin, S.C. Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell. Biol. 1986, 6, 3862–3872. [Google Scholar] [CrossRef]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Cowieson, N.P.; Partridge, J.F.; Allshire, R.C.; McLaughlin, P.J. Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr. Biol. 2000, 10, 517–525. [Google Scholar] [CrossRef]
- Eskeland, R.; Eberharter, A.; Imhof, A. HP1 binding to chromatin methylated at H3K9 is enhanced by auxiliary factors. Mol. Cell. Biol. 2007, 27, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermaak, D.; Malik, H.S. Multiple roles for heterochromatin protein 1 genes in Drosophila. Annu. Rev. Genet. 2009, 43, 467–492. [Google Scholar] [CrossRef]
- Smothers, J.F.; Henikoff, S. The hinge and chromo shadow domain impart distinct targeting of HP1-like proteins. Mol. Cell. Biol. 2001, 21, 2555–2569. [Google Scholar] [CrossRef] [Green Version]
- Eissenberg, J.C.; Reuter, G. Cellular mechanism for targeting heterochromatin formation in Drosophila. Int. Rev. Cell Mol. Biol. 2009, 273, 1–47. [Google Scholar]
- Maison, C.; Almouzni, G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004, 5, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Vicient, C.M.; Casacuberta, J.M. Impact of transposable elements on polyploid plant genomes. Ann. Bot. 2017, 120, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Heun, P. Nucleolus and nuclear periphery: Velcro for heterochromatin. Curr. Opin. Cell Biol. 2014, 28, 54–60. [Google Scholar] [CrossRef]
- Minc, E.; Allory, Y.; Worman, H.J.; Courvalin, J.C.; Buendia, B. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 1999, 108, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Wreggett, K.A.; Hill, F.; James, P.S.; Hutchings, A.; Butcher, G.W.; Singh, P.B. A mammalian homologue of Drosophila heterochromatin protein 1 (HP1) is a component of constitutive heterochromatin. Cytogenet. Cell Genet. 1994, 66, 99–103. [Google Scholar] [CrossRef]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schöfer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster-Böckler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef]
- Cramer, D.; Serrano, L.; Schaefer, M.H. A network of epigenetic modifiers and DNA repair genes controls tissue-specific copy number alteration preference. Elife 2016, 5, e16519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.; Medema, R.H. Genetic instability: Tipping the balance. Oncogene 2013, 32, 4459–4470. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.C.; Karpen, G.H. Epigenetic regulation of heterochromatic DNA stability. Curr. Opin. Genet. Dev. 2008, 18, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Roukos, V.; Misteli, T. The biogenesis of chromosome translocations. Nat. Cell Biol. 2014, 16, 293–300. [Google Scholar] [CrossRef]
- Torres-Rosell, J.; Sunjevaric, I.; De Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragon, L.; Lisby, M. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef]
- Chiolo, I.; Tang, J.; Georgescu, W.; Costes, S.V. Nuclear dynamics of radiation-induced foci in euchromatin and heterochromatin. Mutat. Res. 2013, 750, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [Green Version]
- Zeller, P.; Padeken, J.; van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef]
- Montavon, T.; Shukeir, N.; Erikson, G.; Engist, B.; Onishi-Seebacher, M.; Ryan, D.; Musa, Y.; Mittler, G.; Meyer, A.G.; Genoud, C.; et al. Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat. Commun. 2021, 12, 4359. [Google Scholar] [CrossRef]
- Peng, J.C.; Karpen, G.H. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat. Cell Biol. 2007, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Karpen, G.H. Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS Genet. 2009, 5, e1000435. [Google Scholar] [CrossRef] [Green Version]
- Padeken, J.; Zeller, P.; Towbin, B.; Katic, I.; Kalck, V.; Methot, S.P.; Gasser, S.M. Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev. 2019, 33, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noon, A.T.; Shibata, A.; Rief, N.; Löbrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM Signaling Facilitates Repair of DNA Double-Strand Breaks Associated with Heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Lorković, Z.J.; Park, C.; Goiser, M.; Jiang, D.; Kurzbauer, M.-T.; Schlögelhofer, P.; Berger, F. Compartmentalization of DNA Damage Response between Heterochromatin and Euchromatin Is Mediated by Distinct H2A Histone Variants. Curr. Biol. 2017, 27, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-H.; Paulson, A.; Abmayr, S.M.; Workman, J.L. HP1a targets the Drosophila KDM4A demethylase to a subset of heterochromatic genes to regulate H3K36me3 levels. PLoS ONE 2012, 7, e39758. [Google Scholar] [CrossRef] [Green Version]
- Crona, F.; Dahlberg, O.; Lundberg, L.E.; Larsson, J.; Mannervik, M. Gene regulation by the lysine demethylase KDM4A in Drosophila. Dev. Biol. 2013, 373, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, R.J.; Yamane, K.; Bae, Y.; Zhang, D.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 2006, 442, 312–316. [Google Scholar] [CrossRef]
- Lloret-Llinares, M.; Carré, C.; Vaquero, A.; De Olano, N.; Azorín, F. Characterization of Drosophila melanogaster JmjC+N histone demethylases. Nucleic Acids Res. 2008, 36, 2852–2863. [Google Scholar] [CrossRef] [Green Version]
- Tsurumi, A.; Dutta, P.; Yan, S.-J.; Shang, R.; Li, W.X. Drosophila Kdm4 demethylases in histone H3 lysine 9 demethylation and ecdysteroid signaling. Sci. Rep. 2013, 3, 2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmenares, S.U.; Swenson, J.M.; Langley, S.A.; Kennedy, C.; Costes, S.V.; Karpen, G.H. Drosophila Histone Demethylase KDM4A Has Enzymatic and Non-enzymatic Roles in Controlling Heterochromatin Integrity. Dev. Cell 2017, 42, 156–169.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.; Colmenares, S.U.; Lee, T.; Karpen, G.H. Timely double-strand break repair and pathway choice in pericentromeric heterochromatin depend on the histone demethylase dKDM4A. Genes Dev. 2019, 33, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.C.; Manning, A.L.; Van Rechem, C.; Kim, J.; Ladd, B.; Cho, J.; Pineda, C.M.; Murphy, N.; Daniels, D.L.; Montagna, C.; et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 2013, 154, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury-Haddad, H.; Guttmann-Raviv, N.; Ipenberg, I.; Huggins, D.; Jeyasekharan, A.D.; Ayoub, N. PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E728–E737. [Google Scholar] [CrossRef] [Green Version]
- Garvin, A.J.; Morris, J.R. SUMO, a small, but powerful, regulator of double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160281. [Google Scholar] [CrossRef] [Green Version]
- Aragon, L. The Smc5/6 Complex: New and Old Functions of the Enigmatic Long-Distance Relative. Annu. Rev. Genet. 2018, 52, 89–107. [Google Scholar] [CrossRef]
- Ryu, T.; Bonner, M.R.; Chiolo, I. Cervantes and Quijote protect heterochromatin from aberrant recombination and lead the way to the nuclear periphery. Nucleus 2016, 7, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef]
- Caridi, C.P.; Plessner, M.; Grosse, R.; Chiolo, I. Nuclear actin filaments in DNA repair dynamics. Nat. Cell Biol. 2019, 21, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef]
- Geli, V.; Lisby, M. Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays 2015, 37, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Amaral, N.; Ryu, T.; Li, X.; Chiolo, I. Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 2017, 33, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Freudenreich, C.H.; Su, X.A. Relocalization of DNA lesions to the nuclear pore complex. FEMS Yeast Res. 2016, 16, fow095. [Google Scholar] [CrossRef]
- Nagai, S.; Dubrana, K.; Tsai-Pflugfelder, M.; Davidson, M.B.; Roberts, T.M.; Brown, G.W.; Varela, E.; Hediger, F.; Gasser, S.M.; Krogan, N.J. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 2008, 322, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzaru, A.M.; Kareh, M.; Lamm, N.; Lazzerini-Denchi, E.; Cesare, A.J.; Sfeir, A. Replication stress conferred by POT1 dysfunction promotes telomere relocalization to the nuclear pore. Genes Dev. 2020, 34, 1619–1636. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Read, M.N.; Nobis, M.; Van Ly, D.; Page, S.G.; Masamsetti, V.P.; Timpson, P.; Biro, M.; Cesare, A.J. Nuclear F-actin counteracts nuclear deformation and promotes fork repair during replication stress. Nat. Cell Biol. 2020, 22, 1460–1470. [Google Scholar] [CrossRef]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, R.; Tones, J.; Liu, M.; Dilley, R.L.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 2020, 31, 2048–2056. [Google Scholar] [CrossRef]
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590. [Google Scholar] [CrossRef]
- Nagai, S.; Davoodi, N.; Gasser, S.M. Nuclear organization in genome stability: SUMO connections. Cell Res. 2011, 21, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Barr, M.L.; Bertram, E.G. A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 1949, 163, 676. [Google Scholar] [CrossRef]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Buchenau, P.; Hodgson, J.; Strutt, H.; Arndt-Jovin, D.J. The distribution of polycomb-group proteins during cell division and development in Drosophila embryos: Impact on models for silencing. J. Cell Biol. 1998, 141, 469–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saurin, A.J.; Shiels, C.; Williamson, J.; Satijn, D.P.; Otte, A.P.; Sheer, D.; Freemont, P.S. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J. Cell Biol. 1998, 142, 887–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantignies, F.; Roure, V.; Comet, I.; Leblanc, B.; Schuettengruber, B.; Bonnet, J.; Tixier, V.; Mas, A.; Cavalli, G. Polycomb-dependent regulatory contacts between distant HOX loci in Drosophila. Cell 2011, 144, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Lanzuolo, C.; Roure, V.; Dekker, J.; Bantignies, F.; Orlando, V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat. Cell Biol. 2007, 9, 1167–1174. [Google Scholar] [CrossRef]
- Lewis, P.H. Pc: Polycomb. Drosoph. Inh. Serv. 1947, 21, 69. [Google Scholar]
- Duncan, I.M. Polycomblike: A gene that appears to be required for the normal expression of the bithorax and antennapedia gene complexes of Drosophila melanogaster. Genetics 1982, 102, 49–70. [Google Scholar] [CrossRef]
- Jürgens, G. A group of genes controlling the spatial expression of the bithorax complex in Drosophila. Nature 1985, 316, 153–155. [Google Scholar] [CrossRef]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Pengelly, A.R.; Copur, Ö.; Jäckle, H.; Herzig, A.; Müller, J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 2013, 339, 698–699. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Raible, F.; Mollaaghababa, R.; Guyon, J.R.; Wu, C.T.; Bender, W.; Kingston, R.E. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 1999, 98, 37–46. [Google Scholar] [CrossRef] [Green Version]
- King, I.F.; Emmons, R.B.; Francis, N.J.; Wild, B.; Müller, J.; Kingston, R.E.; Wu, C.T. Analysis of a polycomb group protein defines regions that link repressive activity on nucleosomal templates to in vivo function. Mol. Cell. Biol. 2005, 25, 6578–6591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin compaction by a polycomb group protein complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef] [Green Version]
- Grau, D.J.; Chapman, B.A.; Garlick, J.D.; Borowsky, M.; Francis, N.J.; Kingston, R.E. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 2011, 25, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Lau, M.S.; Schwartz, M.G.; Kundu, S.; Savol, A.J.; Wang, P.I.; Marr, S.K.; Grau, D.J.; Schorderet, P.; Sadreyev, R.I.; Tabin, C.J.; et al. Mutation of a nucleosome compaction region disrupts Polycomb-mediated axial patterning. Science 2017, 355, 1081–1084. [Google Scholar] [CrossRef] [Green Version]
- Seif, E.; Kang, J.J.; Sasseville, C.; Senkovich, O.; Kaltashov, A.; Boulier, E.L.; Kapur, I.; Kim, C.A.; Francis, N.J. Phase separation by the polyhomeotic sterile alpha motif compartmentalizes Polycomb Group proteins and enhances their activity. Nat. Commun. 2020, 11, 5609. [Google Scholar] [CrossRef]
- Das, P.; Taube, J.H. Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity. Cancers 2020, 12, 2792. [Google Scholar] [CrossRef]
- Müller, I.; Merk, B.; Voss, K.O.; Averbeck, N.; Jakob, B.; Durante, M.; Taucher-Scholz, G. Species conserved DNA damage response at the inactive human X chromosome. Mutat. Res. 2013, 756, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Kallimasioti-Paz, E.M.; Thelakkad Chathoth, K.; Taylor, G.C.; Meynert, A.; Ballinger, T.; Kelder, M.J.E.; Lalevée, S.; Sanli, I.; Feil, R.; Wood, A.J. Heterochromatin delays CRISPR-Cas9 mutagenesis but does not influence the outcome of mutagenic DNA repair. PLoS Biol. 2018, 16, e2005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, J.R.; Bartolomei, M.S. Chromatin regulators of genomic imprinting. Biochim. Biophys. Acta 2014, 1839, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; van der Weide, R.H.; Morris, B.; van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; van den Berg, J.; et al. Impact of chromatin context on Cas9-induced DNA double-strand break repair pathway balance. Mol. Cell 2021, 81, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Fitieh, A.; Locke, A.J.; Motamedi, M.; Ismail, I.H. The Role of Polycomb Group Protein BMI1 in DNA Repair and Genomic Stability. Int. J. Mol. Sci. 2021, 22, 2976. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef]
- Pickersgill, H.; Kalverda, B.; de Wit, E.; Talhout, W.; Fornerod, M.; van Steensel, B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 2006, 38, 1005–1014. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Ikegami, K.; Egelhofer, T.A.; Strome, S.; Lieb, J.D. Caenorhabditis elegans chromosome arms are anchored to the nuclear membrane via discontinuous association with LEM-2. Genome Biol. 2010, 11, R120. [Google Scholar] [CrossRef] [Green Version]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaitre, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear position dictates DNA repair pathway choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef] [Green Version]
- Politz, J.C.R.; Scalzo, D.; Groudine, M. The redundancy of the mammalian heterochromatic compartment. Curr. Opin. Genet. Dev. 2016, 37, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, B.A.; Karpen, G.H. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 2004, 11, 1076–1083. [Google Scholar] [CrossRef]
- Warmerdam, D.O.; van den Berg, J.; Medema, R.H. Breaks in the 45S rDNA Lead to Recombination-Mediated Loss of Repeats. Cell Rep. 2016, 14, 2519–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Sluis, M.; McStay, B. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015, 29, 1151–1163. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Boiarsky, J.A.; Greenberg, R.A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 2015, 13, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef] [Green Version]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delabaere, L.; Ertl, H.A.; Massey, D.J.; Hofley, C.M.; Sohail, F.; Bienenstock, E.J.; Sebastian, H.; Chiolo, I.; LaRocque, J.R. Aging impairs double-strand break repair by homologous recombination in Drosophila germ cells. Aging Cell 2017, 16, 320–328. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kendek, A.; Wensveen, M.R.; Janssen, A. The Sound of Silence: How Silenced Chromatin Orchestrates the Repair of Double-Strand Breaks. Genes 2021, 12, 1415. https://doi.org/10.3390/genes12091415
Kendek A, Wensveen MR, Janssen A. The Sound of Silence: How Silenced Chromatin Orchestrates the Repair of Double-Strand Breaks. Genes. 2021; 12(9):1415. https://doi.org/10.3390/genes12091415
Chicago/Turabian StyleKendek, Apfrida, Marieke R. Wensveen, and Aniek Janssen. 2021. "The Sound of Silence: How Silenced Chromatin Orchestrates the Repair of Double-Strand Breaks" Genes 12, no. 9: 1415. https://doi.org/10.3390/genes12091415