
3D Genome Organization: Causes and Consequences for DNA Damage and Repair


{kind=link}
{kind=link}
{kind=link}
Abstract
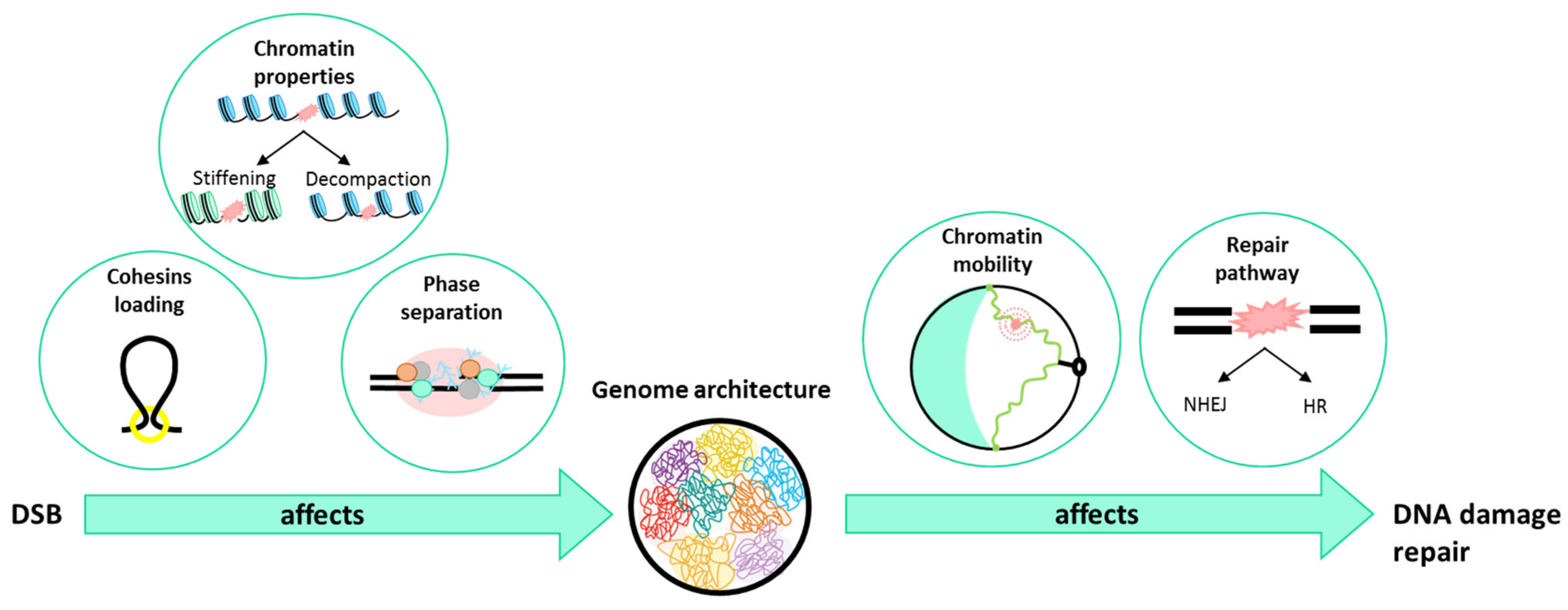
:1. Introduction
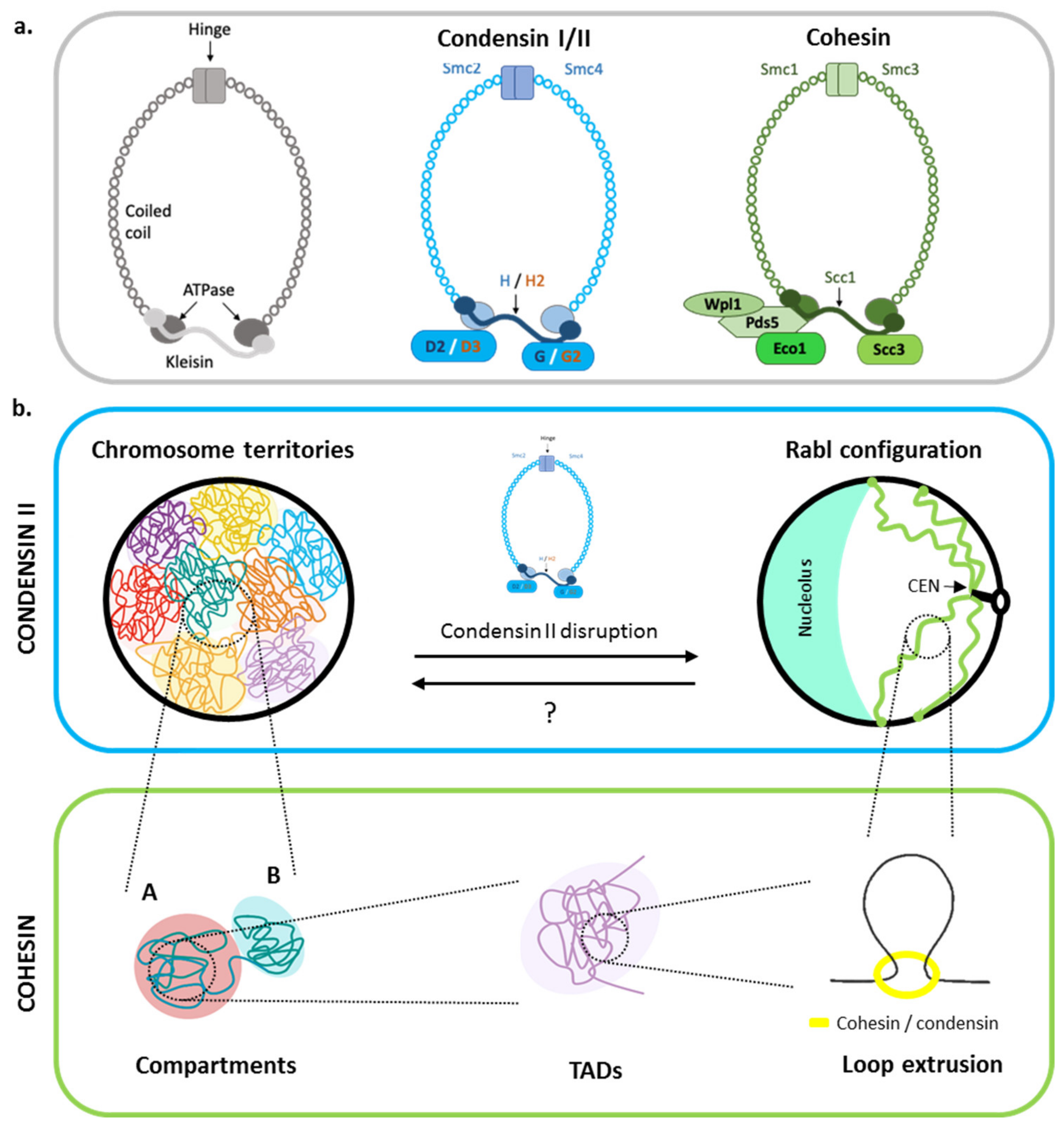
2. Genome Organization without Damage: The Interplay between the Chromatin Scales
2.1. Chromosome Territories and Rabl-like Chromosome Configurations
2.2. The Role of Cohesins in Chromosome Compartment Organization

2.3. Loop Extrusion
2.4. Nuclear Envelope Tethering: A Prominent Role in Genome Organization
3. Are Some Genomic Regions More Fragile Than Others?
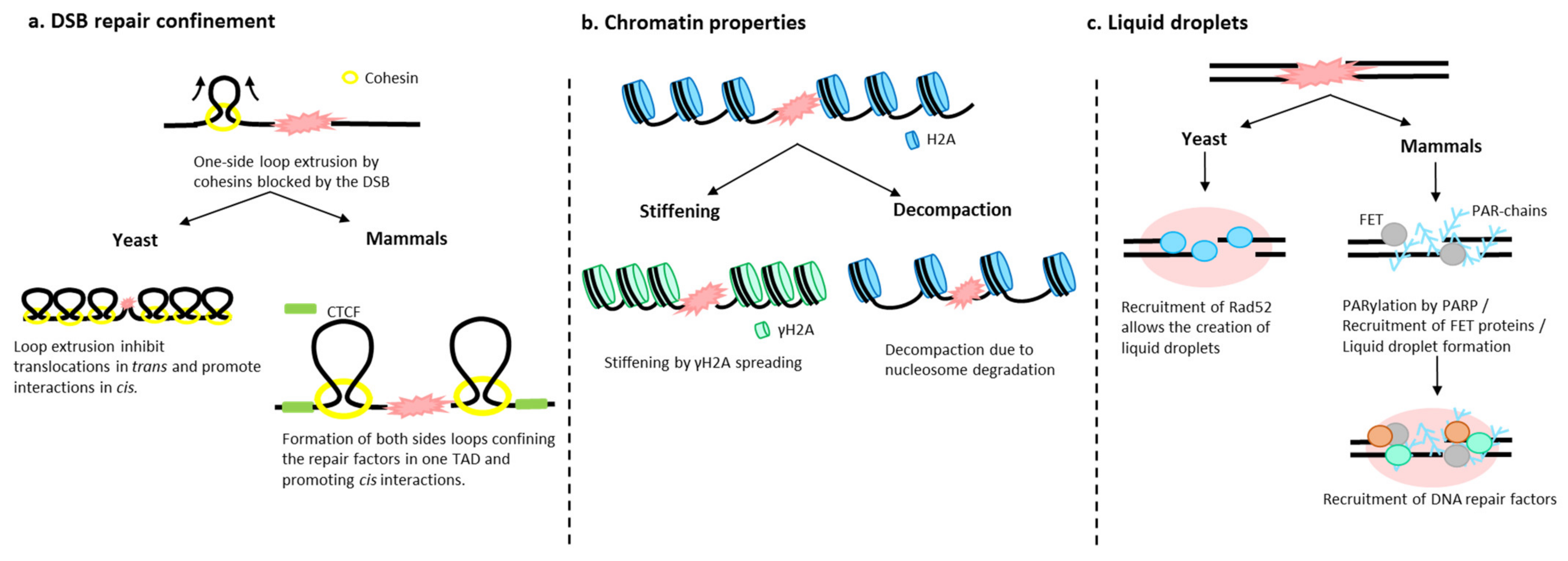
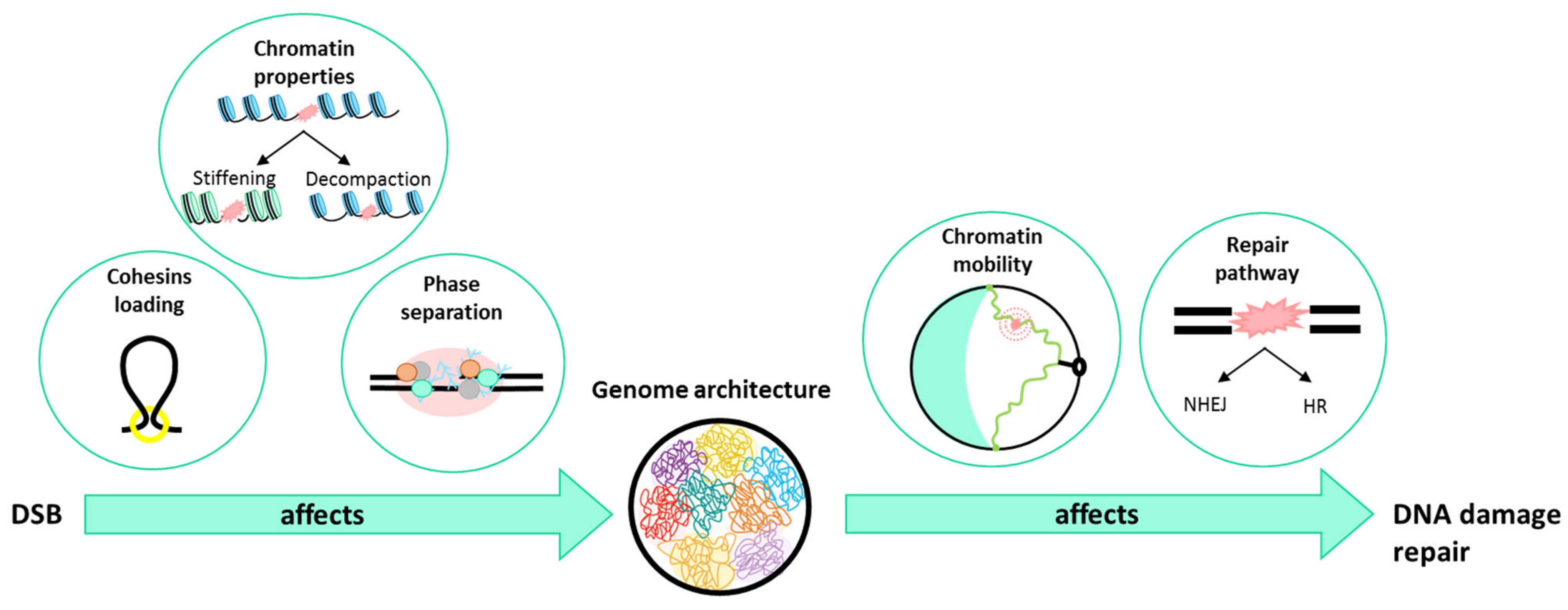
4. What Is Happening to the Chromatin When a DSB Occurs?
4.1. Chromatin Architecture Changes: Role of Cohesins
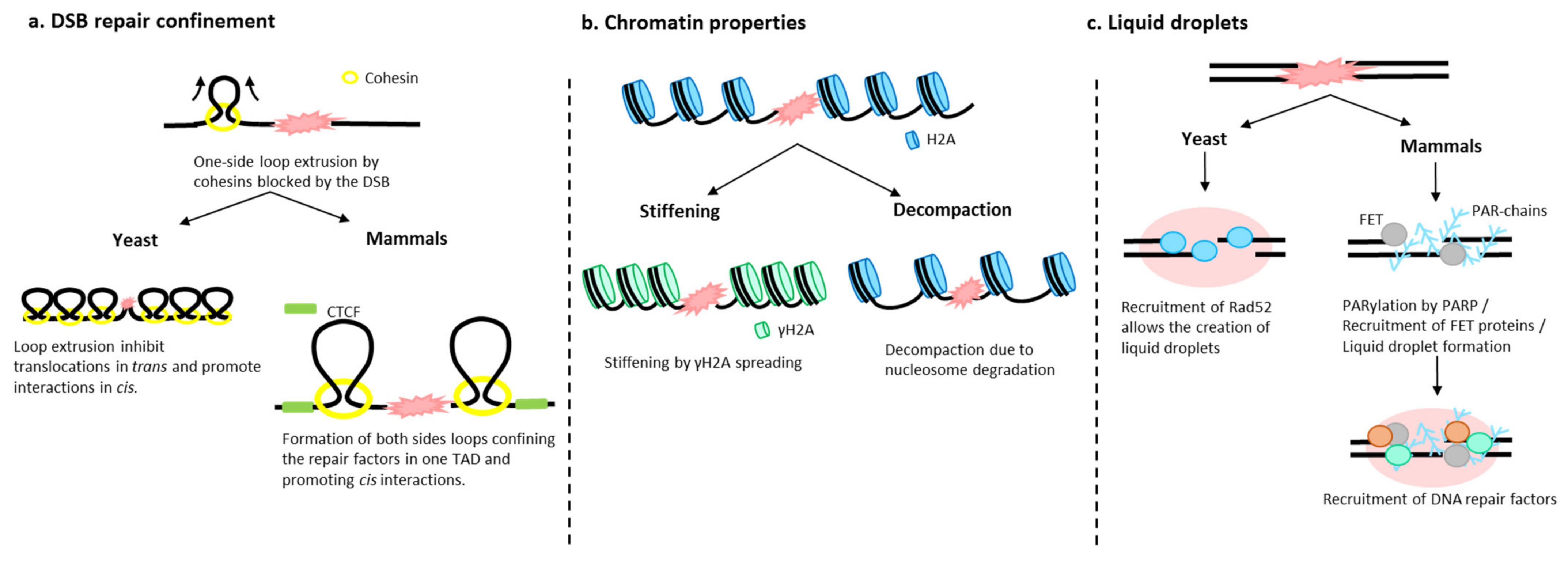
4.2. Chromatin Fiber Changes upon DSB
4.3. Role of Liquid–Liquid Phase in DSB Foci
5. How Does Genome Organization Affect Repair?
5.1. In the Choice of the Repair Pathway
5.2. In Chromatin Mobility
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mirny, L.; Dekker, J. Mechanisms of Chromosome Folding and Nuclear Organization: Their Interplay and Open Questions. Cold Spring Harb. Perspect. Biol. 2021, a040147. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Taddei, A.; Schober, H.; Gasser, S.M. The Budding Yeast Nucleus. Cold Spring Harb. Perspect. Biol. 2010, 2, a000612. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C.; Fabre, E. Principles of Chromosomal Organization: Lessons from Yeast. J. Cell Biol. 2011, 192, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Aragón, L. The Smc5/6 Complex: New and Old Functions of the Enigmatic Long-Distance Relative. Annu. Rev. Genet. 2018, 52, 89–107. [Google Scholar] [CrossRef]
- Hirano, T. Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 2016, 164, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Nasmyth, K.; Haering, C.H. Cohesin: Its Roles and Mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Branco, M.R.; Pombo, A. Intermingling of Chromosome Territories in Interphase Suggests Role in Translocations and Transcription-Dependent Associations. PLoS Biol. 2006, 4, e138. [Google Scholar] [CrossRef] [Green Version]
- Cremer, T.; Cremer, M. Chromosome Territories. Cold Spring Harb. Perspect. Biol. 2010, 2, a003889. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Trelles-Sticken, E.; Scherthan, H.; Loidl, J. Yeast Nuclei Display Prominent Centromere Clustering That Is Reduced in Nondividing Cells and in Meiotic Prophase. J. Cell Biol. 1998, 141, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Schober, H.; Kalck, V.; Vega-Palas, M.A.; Van Houwe, G.; Sage, D.; Unser, M.; Gartenberg, M.R.; Gasser, S.M. Controlled Exchange of Chromosomal Arms Reveals Principles Driving Telomere Interactions in Yeast. Genome Res. 2008, 18, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Therizols, P.; Fairhead, C.; Cabal, G.G.; Genovesio, A.; Olivo-Marin, J.-C.; Dujon, B.; Fabre, E. Telomere Tethering at the Nuclear Periphery Is Essential for Efficient DNA Double Strand Break Repair in Subtelomeric Region. J. Cell Biol. 2006, 172, 189–199. [Google Scholar] [CrossRef]
- Funabiki, H.; Hagan, I.; Uzawa, S.; Yanagida, M. Cell Cycle-Dependent Specific Positioning and Clustering of Centromeres and Telomeres in Fission Yeast. J. Cell Biol. 1993, 121, 961–976. [Google Scholar] [CrossRef] [Green Version]
- Agard, D.A.; Sedat, J.W. Three-Dimensional Architecture of a Polytene Nucleus. Nature 1983, 302, 676–681. [Google Scholar] [CrossRef]
- Hochstrasser, M.; Mathog, D.; Gruenbaum, Y.; Saumweber, H.; Sedat, J.W. Spatial Organization of Chromosomes in the Salivary Gland Nuclei of Drosophila Melanogaster. J. Cell Biol. 1986, 102, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Spector, D.L. Centromere Positioning and Dynamics in Living Arabidopsis Plants. Mol. Biol. Cell 2005, 16, 5710–5718. [Google Scholar] [CrossRef] [Green Version]
- Hoencamp, C.; Dudchenko, O.; Elbatsh, A.M.O.; Brahmachari, S.; Raaijmakers, J.A.; Schaik, T.V.; Cacciatore, Á.S.; Contessoto, V.G.; Heesbeen, R.G.H.P.V.; van den Broek, B.; et al. 3D Genomics across the Tree of Life Reveals Condensin II as a Determinant of Architecture Type. Science 2021, 372, 984–989. [Google Scholar] [CrossRef]
- Litwin, I.; Wysocki, R. New Insights into Cohesin Loading. Curr. Genet. 2018, 64, 53–61. [Google Scholar] [CrossRef]
- Ono, T.; Losada, A.; Hirano, M.; Myers, M.P.; Neuwald, A.F.; Hirano, T. Differential Contributions of Condensin I and Condensin II to Mitotic Chromosome Architecture in Vertebrate Cells. Cell 2003, 115, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T. Condensins: Universal Organizers of Chromosomes with Diverse Functions. Genes Dev. 2012, 26, 1659–1678. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.R.; Hartl, T.A.; Bosco, G. Condensin II Promotes the Formation of Chromosome Territories by Inducing Axial Compaction of Polyploid Interphase Chromosomes. PLoS Genet. 2012, 8, e1002873. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Haarhuis, J.H.I.; van der Weide, R.H.; Blomen, V.A.; Yáñez-Cuna, J.O.; Amendola, M.; van Ruiten, M.S.; Krijger, P.H.L.; Teunissen, H.; Medema, R.H.; van Steensel, B.; et al. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 2017, 169, 693–707.e14. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.; Mirny, L.; et al. Two Independent Modes of Chromatin Organization Revealed by Cohesin Removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wutz, G.; Várnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically Associating Domains and Chromatin Loops Depend on Cohesin and Are Regulated by CTCF, WAPL, and PDS5 Proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef] [PubMed]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin Organization by an Interplay of Loop Extrusion and Compartmental Segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar-Stefanita, L.; Scolari, V.F.; Mercy, G.; Muller, H.; Guérin, T.M.; Thierry, A.; Mozziconacci, J.; Koszul, R. Cohesins and Condensins Orchestrate the 4D Dynamics of Yeast Chromosomes during the Cell Cycle. EMBO J. 2017, 36, 2684–2697. [Google Scholar] [CrossRef] [PubMed]
- Schalbetter, S.A.; Goloborodko, A.; Fudenberg, G.; Belton, J.-M.; Miles, C.; Yu, M.; Dekker, J.; Mirny, L.; Baxter, J. SMC Complexes Differentially Compact Mitotic Chromosomes According to Genomic Context. Nat. Cell Biol. 2017, 19, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial Partitioning of the Regulatory Landscape of the X-Inactivation Centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the Three-Dimensional Organization of Genomes: Interpreting Chromatin Interaction Data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Symmons, O.; Pan, L.; Remeseiro, S.; Aktas, T.; Klein, F.; Huber, W.; Spitz, F. The Shh Topological Domain Facilitates the Action of Remote Enhancers by Reducing the Effects of Genomic Distances. Dev. Cell 2016, 39, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Eser, U.; Chandler-Brown, D.; Ay, F.; Straight, A.F.; Duan, Z.; Noble, W.S.; Skotheim, J.M. Form and Function of Topologically Associating Genomic Domains in Budding Yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E3061–E3070. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Luis, J.; Lazar-Stefanita, L.; Gutierrez-Escribano, P.; Thierry, A.; Cournac, A.; García, A.; González, S.; Sánchez, M.; Jarmuz, A.; Montoya, A.; et al. FACT Mediates Cohesin Function on Chromatin. Nat. Struct. Mol. Biol. 2019, 26, 970–979. [Google Scholar] [CrossRef]
- Hsieh, T.-H.S.; Weiner, A.; Lajoie, B.; Dekker, J.; Friedman, N.; Rando, O.J. Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 2015, 162, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Filippova, G.; Loukinov, D.; Pugacheva, E.; Chen, Q.; Smith, S.T.; Munhall, A.; Grewe, B.; Bartkuhn, M.; Arnold, R.; et al. CTCF Is Conserved from Drosophila to Humans and Confers Enhancer Blocking of the Fab-8 Insulator. EMBO Rep. 2005, 6, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Cattoni, D.I.; Cardozo Gizzi, A.M.; Georgieva, M.; Di Stefano, M.; Valeri, A.; Chamousset, D.; Houbron, C.; Déjardin, S.; Fiche, J.-B.; González, I.; et al. Single-Cell Absolute Contact Probability Detection Reveals Chromosomes Are Organized by Multiple Low-Frequency yet Specific Interactions. Nat. Commun. 2017, 8, 1753. [Google Scholar] [CrossRef] [Green Version]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.-L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, 1502–1515.e10. [Google Scholar] [CrossRef] [Green Version]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.-M. DNA Loop Extrusion by Human Cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef]
- Kim, Y.; Shi, Z.; Zhang, H.; Finkelstein, I.J.; Yu, H. Human Cohesin Compacts DNA by Loop Extrusion. Science 2019, 366, 1345–1349. [Google Scholar] [CrossRef]
- Costantino, L.; Hsieh, T.-H.S.; Lamothe, R.; Darzacq, X.; Koshland, D. Cohesin Residency Determines Chromatin Loop Patterns. eLife 2020, 9, e59889. [Google Scholar] [CrossRef]
- Dauban, L.; Montagne, R.; Thierry, A.; Lazar-Stefanita, L.; Bastié, N.; Gadal, O.; Cournac, A.; Koszul, R.; Beckouët, F. Regulation of Cohesin-Mediated Chromosome Folding by Eco1 and Other Partners. Mol. Cell 2020, 77, 1279–1293.e4. [Google Scholar] [CrossRef]
- Paldi, F.; Alver, B.; Robertson, D.; Schalbetter, S.A.; Kerr, A.; Kelly, D.A.; Baxter, J.; Neale, M.J.; Marston, A.L. Convergent Genes Shape Budding Yeast Pericentromeres. Nature 2020, 582, 119–123. [Google Scholar] [CrossRef]
- Piazza, A.; Bordelet, H.; Dumont, A.; Thierry, A.; Savocco, J.; Girard, F.; Koszul, R. Cohesin Regulates Homology Search during Recombinational DNA Repair. Nat. Cell Biol. 2021, 23, 1176–1186. [Google Scholar] [CrossRef]
- Tedeschi, A.; Wutz, G.; Huet, S.; Jaritz, M.; Wuensche, A.; Schirghuber, E.; Davidson, I.F.; Tang, W.; Cisneros, D.A.; Bhaskara, V.; et al. Wapl Is an Essential Regulator of Chromatin Structure and Chromosome Segregation. Nature 2013, 501, 564–568. [Google Scholar] [CrossRef]
- Lioy, V.S.; Cournac, A.; Marbouty, M.; Duigou, S.; Mozziconacci, J.; Espéli, O.; Boccard, F.; Koszul, R. Multiscale Structuring of the E. Coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell 2018, 172, 771–783.e18. [Google Scholar] [CrossRef]
- Takemata, N.; Samson, R.Y.; Bell, S.D. Physical and Functional Compartmentalization of Archaeal Chromosomes. Cell 2019, 179, 165–179.e18. [Google Scholar] [CrossRef]
- Marbouty, M.; Le Gall, A.; Cattoni, D.I.; Cournac, A.; Koh, A.; Fiche, J.-B.; Mozziconacci, J.; Murray, H.; Koszul, R.; Nollmann, M. Condensin- and Replication-Mediated Bacterial Chromosome Folding and Origin Condensation Revealed by Hi-C and Super-Resolution Imaging. Mol. Cell 2015, 59, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Van Steensel, B.; Delrow, J.; Henikoff, S. Chromatin Profiling Using Targeted DNA Adenine Methyltransferase. Nat. Genet. 2001, 27, 304–308. [Google Scholar] [CrossRef] [Green Version]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [Green Version]
- Gotta, M.; Laroche, T.; Formenton, A.; Maillet, L.; Scherthan, H.; Gasser, S.M. The Clustering of Telomeres and Colocalization with Rap1, Sir3, and Sir4 Proteins in Wild-Type Saccharomyces Cerevisiae. J. Cell Biol. 1996, 134, 1349–1363. [Google Scholar] [CrossRef] [Green Version]
- Taddei, A.; Hediger, F.; Neumann, F.R.; Bauer, C.; Gasser, S.M. Separation of Silencing from Perinuclear Anchoring Functions in Yeast Ku80, Sir4 and Esc1 Proteins. EMBO J. 2004, 23, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin Drives Compartmentalization of Inverted and Conventional Nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.-R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome Organization Drives Chromosome Fragility. Cell 2017, 170, 507–521.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Nieto, P.E.; Schwartz, E.K.; King, D.A.; Paulsen, J.; Collas, P.; Herrera, R.E.; Morrison, A.J. Carcinogen Susceptibility Is Regulated by Genome Architecture and Predicts Cancer Mutagenesis. EMBO J. 2017, 36, 2829–2843. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Breuer, G.A.; Brinkman, E.K.; van der Meulen, A.I.; Borden, S.V.; van Steensel, B.; Bindra, R.S.; LaRocque, J.R.; Karpen, G.H. A Single Double-Strand Break System Reveals Repair Dynamics and Mechanisms in Heterochromatin and Euchromatin. Genes Dev. 2016, 30, 1645–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Rosell, J.; Sunjevaric, I.; De Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragón, L.; Lisby, M. The Smc5-Smc6 Complex and SUMO Modification of Rad52 Regulates Recombinational Repair at the Ribosomal Gene Locus. Nat. Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef]
- Bouwman, B.A.M.; Crosetto, N. Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling. Genes 2018, 9, 632. [Google Scholar] [CrossRef] [Green Version]
- Baranello, L.; Kouzine, F.; Wojtowicz, D.; Cui, K.; Przytycka, T.M.; Zhao, K.; Levens, D. DNA Break Mapping Reveals Topoisomerase II Activity Genome-Wide. Int. J. Mol. Sci. 2014, 15, 13111–13122. [Google Scholar] [CrossRef] [Green Version]
- Mourad, R.; Ginalski, K.; Legube, G.; Cuvier, O. Predicting Double-Strand DNA Breaks Using Epigenome Marks or DNA at Kilobase Resolution. Genome Biol. 2018, 19, 34. [Google Scholar] [CrossRef] [Green Version]
- Gothe, H.J.; Bouwman, B.A.M.; Gusmao, E.G.; Piccinno, R.; Petrosino, G.; Sayols, S.; Drechsel, O.; Minneker, V.; Josipovic, N.; Mizi, A.; et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol. Cell 2019, 75, 267–283.e12. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Huang, S.-Y.N.; Wutz, G.; Tang, W.; Zagnoli-Vieira, G.; Callen, E.; Wong, N.; Day, A.; Peters, J.-M.; et al. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol. Cell 2019, 75, 252–266.e8. [Google Scholar] [CrossRef]
- Callahan, J.L.; Andrews, K.J.; Zakian, V.A.; Freudenreich, C.H. Mutations in Yeast Replication Proteins That Increase CAG/CTG Expansions Also Increase Repeat Fragility. Mol. Cell. Biol. 2003, 23, 7849–7860. [Google Scholar] [CrossRef] [Green Version]
- Lobachev, K.S.; Gordenin, D.A.; Resnick, M.A. The Mre11 Complex Is Required for Repair of Hairpin-Capped Double-Strand Breaks and Prevention of Chromosome Rearrangements. Cell 2002, 108, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Samadashwily, G.M.; Raca, G.; Mirkin, S.M. Trinucleotide Repeats Affect DNA Replication in Vivo. Nat. Genet. 1997, 17, 298–304. [Google Scholar] [CrossRef]
- Kim, J.-S.; Krasieva, T.B.; LaMorte, V.; Taylor, A.M.R.; Yokomori, K. Specific Recruitment of Human Cohesin to Laser-Induced DNA Damage. J. Biol. Chem. 2002, 277, 45149–45153. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-T.; Xu, B.; Kastan, M.B. Involvement of the Cohesin Protein, Smc1, in Atm-Dependent and Independent Responses to DNA Damage. Genes Dev. 2002, 16, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-S.; Lee, K.; Legube, G.; Haber, J.E. Dynamics of Yeast Histone H2A and H2B Phosphorylation in Response to a Double-Strand Break. Nat. Struct. Mol. Biol. 2014, 21, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and Dynamics of Chromatin Modification Induced by a Defined DNA Double-Strand Break. Curr. Biol. 2004, 14, 1703–1711. [Google Scholar] [CrossRef] [Green Version]
- Ström, L.; Sjögren, C. DNA Damage-Induced Cohesion. Cell Cycle Georget. Tex 2005, 4, 536–539. [Google Scholar] [CrossRef] [Green Version]
- Ünal, E.; Arbel-Eden, A.; Sattler, U.; Shroff, R.; Lichten, M.; Haber, J.E.; Koshland, D. DNA Damage Response Pathway Uses Histone Modification to Assemble a Double-Strand Break-Specific Cohesin Domain. Mol. Cell 2004, 16, 991–1002. [Google Scholar] [CrossRef]
- Arnould, C.; Rocher, V.; Finoux, A.-L.; Clouaire, T.; Li, K.; Zhou, F.; Caron, P.; Mangeot, P.E.; Ricci, E.P.; Mourad, R.; et al. Loop Extrusion as a Mechanism for Formation of DNA Damage Repair Foci. Nature 2021, 590, 660–665. [Google Scholar] [CrossRef]
- Betts Lindroos, H.; Ström, L.; Itoh, T.; Katou, Y.; Shirahige, K.; Sjögren, C. Chromosomal Association of the Smc5/6 Complex Reveals That It Functions in Differently Regulated Pathways. Mol. Cell 2006, 22, 755–767. [Google Scholar] [CrossRef]
- De Piccoli, G.; Cortes-Ledesma, F.; Ira, G.; Torres-Rosell, J.; Uhle, S.; Farmer, S.; Hwang, J.-Y.; Machin, F.; Ceschia, A.; McAleenan, A.; et al. Smc5–Smc6 Mediate DNA Double-Strand-Break Repair by Promoting Sister-Chromatid Recombination. Nat. Cell Biol. 2006, 8, 1032–1034. [Google Scholar] [CrossRef]
- Potts, P.R.; Porteus, M.H.; Yu, H. Human SMC5/6 Complex Promotes Sister Chromatid Homologous Recombination by Recruiting the SMC1/3 Cohesin Complex to Double-Strand Breaks. EMBO J. 2006, 25, 3377–3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onoda, F.; Takeda, M.; Seki, M.; Maeda, D.; Tajima, J.; Ui, A.; Yagi, H.; Enomoto, T. SMC6 Is Required for MMS-Induced Interchromosomal and Sister Chromatid Recombinations in Saccharomyces Cerevisiae. DNA Repair 2004, 3, 429–439. [Google Scholar] [CrossRef]
- Torres-Rosell, J.; Machín, F.; Farmer, S.; Jarmuz, A.; Eydmann, T.; Dalgaard, J.Z.; Aragón, L. SMC5 and SMC6 Genes Are Required for the Segregation of Repetitive Chromosome Regions. Nat. Cell Biol. 2005, 7, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Herbert, S.; Brion, A.; Arbona, J.; Lelek, M.; Veillet, A.; Lelandais, B.; Parmar, J.; Fernández, F.G.; Almayrac, E.; Khalil, Y.; et al. Chromatin Stiffening Underlies Enhanced Locus Mobility after DNA Damage in Budding Yeast. EMBO J. 2017, 36, 2595–2608. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Bustamante, C. Pulling a Single Chromatin Fiber Reveals the Forces That Maintain Its Higher-Order Structure. Proc. Natl. Acad. Sci. USA 2000, 97, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miné-Hattab, J.; Recamier, V.; Izeddin, I.; Rothstein, R.; Darzacq, X. Multi-Scale Tracking Reveals Scale-Dependent Chromatin Dynamics after DNA Damage. Mol. Biol. Cell 2017, 28, 3323–3332. [Google Scholar] [CrossRef]
- Burgess, R.C.; Burman, B.; Kruhlak, M.; Misteli, T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Hauer, M.H.; Seeber, A.; Singh, V.; Thierry, R.; Sack, R.; Amitai, A.; Kryzhanovska, M.; Eglinger, J.; Holcman, D.; Owen-Hughes, T.; et al. Histone Degradation in Response to DNA Damage Enhances Chromatin Dynamics and Recombination Rates. Nat. Struct. Mol. Biol. 2017, 24, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Cheblal, A.; Challa, K.; Seeber, A.; Shimada, K.; Yoshida, H.; Ferreira, H.C.; Amitai, A.; Gasser, S.M. DNA Damage-Induced Nucleosome Depletion Enhances Homology Search Independently of Local Break Movement. Mol. Cell 2020, 80, 311–326.e4. [Google Scholar] [CrossRef]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.-B.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid Demixing of Intrinsically Disordered Proteins Is Seeded by Poly(ADP-Ribose). Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Oshidari, R.; Huang, R.; Medghalchi, M.; Tse, E.Y.W.; Ashgriz, N.; Lee, H.O.; Wyatt, H.; Mekhail, K. DNA Repair by Rad52 Liquid Droplets. Nat. Commun. 2020, 11, 695. [Google Scholar] [CrossRef] [Green Version]
- Miné-Hattab, J.; Heltberg, M.; Villemeur, M.; Guedj, C.; Mora, T.; Walczak, A.M.; Dahan, M.; Taddei, A. Single Molecule Microscopy Reveals Key Physical Features of Repair Foci in Living Cells. eLife 2021, 10, e60577. [Google Scholar] [CrossRef]
- García Fernández, F.; Lemos, B.; Khalil, Y.; Batrin, R.; Haber, J.E.; Fabre, E. Modified Chromosome Structure Caused by Phosphomimetic H2A Modulates the DNA Damage Response by Increasing Chromatin Mobility in Yeast. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef]
- Lisby, M.; Rothstein, R.; Mortensen, U.H. Rad52 Forms DNA Repair and Recombination Centers during S Phase. Proc. Natl. Acad. Sci. USA 2001, 98, 8276–8282. [Google Scholar] [CrossRef] [Green Version]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-Wide Mapping of Long-Range Contacts Unveils Clustering of DNA Double-Strand Breaks at Damaged Active Genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Clouaire, T.; Legube, G. DNA Double Strand Break Repair Pathway Choice: A Chromatin Based Decision? Nucleus 2015, 6, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Kalousi, A.; Soutoglou, E. Nuclear Compartmentalization of DNA Repair. Curr. Opin. Genet. Dev. 2016, 37, 148–157. [Google Scholar] [CrossRef]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in Genome Editing through Control of DNA Repair Pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldeyron, C.; Soria, G.; Roche, D.; Cook, A.J.L.; Almouzni, G. HP1α Recruitment to DNA Damage by P150CAF-1 Promotes Homologous Recombination Repair. J. Cell Biol. 2011, 193, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; van der Weide, R.H.; Morris, B.; van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; van den Berg, J.; et al. Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 2021, 81, 2216–2230.e10. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear Position Dictates DNA Repair Pathway Choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef] [Green Version]
- Nagai, S.; Dubrana, K.; Tsai-Pflugfelder, M.; Davidson, M.B.; Roberts, T.M.; Brown, G.W.; Varela, E.; Hediger, F.; Gasser, S.M.; Krogan, N.J. Functional Targeting of DNA Damage to a Nuclear Pore-Associated SUMO-Dependent Ubiquitin Ligase. Science 2008, 322, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Oza, P.; Jaspersen, S.L.; Miele, A.; Dekker, J.; Peterson, C.L. Mechanisms That Regulate Localization of a DNA Double-Strand Break to the Nuclear Periphery. Genes Dev. 2009, 23, 912–927. [Google Scholar] [CrossRef] [Green Version]
- Agmon, N.; Liefshitz, B.; Zimmer, C.; Fabre, E.; Kupiec, M. Effect of Nuclear Architecture on the Efficiency of Double-Strand Break Repair. Nat. Cell Biol. 2013, 15, 694–699. [Google Scholar] [CrossRef]
- Lee, C.-S.; Wang, R.W.; Chang, H.-H.; Capurso, D.; Segal, M.R.; Haber, J.E. Chromosome Position Determines the Success of Double-Strand Break Repair. Proc. Natl. Acad. Sci. USA 2016, 113, E146–E154. [Google Scholar] [CrossRef] [Green Version]
- Miné-Hattab, J.; Taddei, A. Physical Principles and Functional Consequences of Nuclear Compartmentalization in Budding Yeast. Curr. Opin. Cell Biol. 2019, 58, 105–113. [Google Scholar] [CrossRef]
- Lottersberger, F.; Karssemeijer, R.A.; Dimitrova, N.; de Lange, T. 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell 2015, 163, 880–893. [Google Scholar] [CrossRef] [Green Version]
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial Dynamics of Chromosome Translocations in Living Cells. Science 2013, 341, 660–664. [Google Scholar] [CrossRef]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA Double-Strand Breaks Revealed by Clustering of Damaged Chromosome Domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Spichal, M.; Brion, A.; Herbert, S.; Cournac, A.; Marbouty, M.; Zimmer, C.; Koszul, R.; Fabre, E. Evidence for a Dual Role of Actin in Regulating Chromosome Organization and Dynamics in Yeast. J. Cell Sci. 2016, 129, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Lawrimore, J.; Barry, T.M.; Barry, R.M.; York, A.C.; Friedman, B.; Cook, D.M.; Akialis, K.; Tyler, J.; Vasquez, P.; Yeh, E.; et al. Microtubule Dynamics Drive Enhanced Chromatin Motion and Mobilize Telomeres in Response to DNA Damage. Mol. Biol. Cell 2017, 28, 1701–1711. [Google Scholar] [CrossRef]
- Strecker, J.; Gupta, G.D.; Zhang, W.; Bashkurov, M.; Landry, M.-C.; Pelletier, L.; Durocher, D. DNA Damage Signalling Targets the Kinetochore to Promote Chromatin Mobility. Nat. Cell Biol. 2016, 18, 281–290. [Google Scholar] [CrossRef]
- Heun, P.; Laroche, T.; Shimada, K.; Furrer, P.; Gasser, S.M. Chromosome Dynamics in the Yeast Interphase Nucleus. Science 2001, 294, 2181–2186. [Google Scholar] [CrossRef]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-Strand Breaks in Heterochromatin Move Outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-Actin and Myosins Drive Relocalization of Heterochromatic Breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef]
- Dion, V.; Kalck, V.; Seeber, A.; Schleker, T.; Gasser, S.M. Cohesin and the Nucleolus Constrain the Mobility of Spontaneous Repair Foci. EMBO Rep. 2013, 14, 984–991. [Google Scholar] [CrossRef] [Green Version]
- Van der Crabben, S.N.; Hennus, M.P.; McGregor, G.A.; Ritter, D.I.; Nagamani, S.C.S.; Wells, O.S.; Harakalova, M.; Chinn, I.K.; Alt, A.; Vondrova, L.; et al. Destabilized SMC5/6 Complex Leads to Chromosome Breakage Syndrome with Severe Lung Disease. J. Clin. Investig. 2016, 126, 2881–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Krantz, I. Cornelia de Lange Syndrome, Cohesin, and Beyond. Clin. Genet. 2009, 76, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carré-Simon, À.; Fabre, E. 3D Genome Organization: Causes and Consequences for DNA Damage and Repair. Genes 2022, 13, 7. https://doi.org/10.3390/genes13010007
Carré-Simon À, Fabre E. 3D Genome Organization: Causes and Consequences for DNA Damage and Repair. Genes. 2022; 13(1):7. https://doi.org/10.3390/genes13010007
Chicago/Turabian StyleCarré-Simon, Ànnia, and Emmanuelle Fabre. 2022. "3D Genome Organization: Causes and Consequences for DNA Damage and Repair" Genes 13, no. 1: 7. https://doi.org/10.3390/genes13010007
APA StyleCarré-Simon, À., & Fabre, E. (2022). 3D Genome Organization: Causes and Consequences for DNA Damage and Repair. Genes, 13(1), 7. https://doi.org/10.3390/genes13010007