


Gene Panel Sequencing Identifies a Novel RYR1 p.Ser2300Pro Variant as Candidate for Malignant Hyperthermia with Multi-Minicore Myopathy

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Muscle Biopsy
2.2. Succinate Dehydrogenase Staining
2.3. Gene Panel Sequencing
2.4. Sanger Sequencing
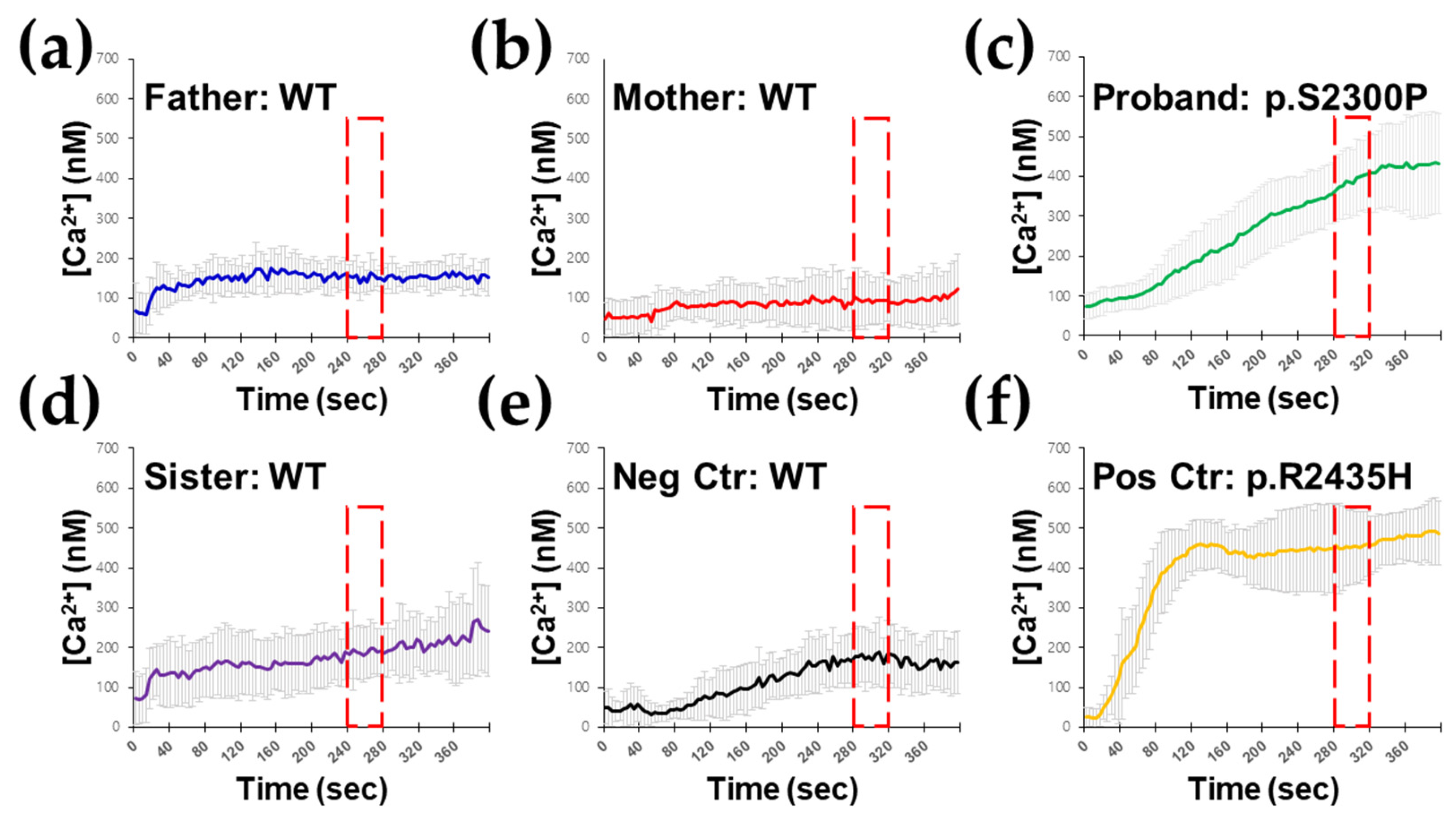
2.5. Ex Vivo RYR1-Mediated Intracellular Ca2+ Release Testing in B Lymphocytes
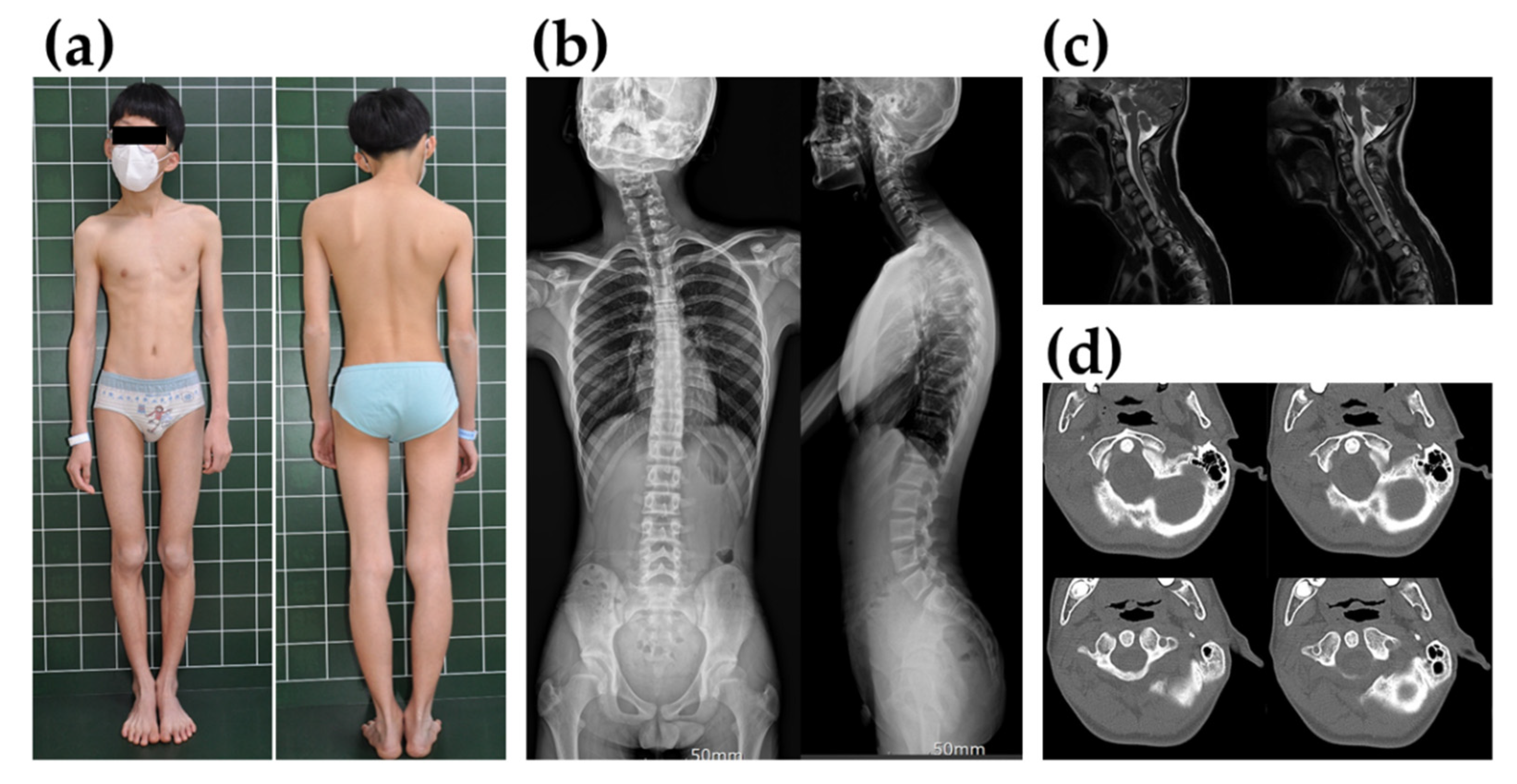
3. Case Presentation
4. Results
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosero, E.B.; Adesanya, A.O.; Timaran, C.H.; Joshi, G.P. Trends and outcomes of malignant hyperthermia in the United States, 2000 to 2005. Anesthesiology 2009, 110, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.; Pollock, N.; Schiemann, A.; Bulger, T.; Stowell, K. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 2015, 10, 93. [Google Scholar] [CrossRef]
- Mickelson, J.R.; Louis, C.F. Malignant hyperthermia: Excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol. Rev. 1996, 76, 537–592. [Google Scholar] [CrossRef] [PubMed]
- Duke, A.M.; Hopkins, P.M.; Calaghan, S.C.; Halsall, J.P.; Steele, D.S. Store-operated Ca2+ entry in malignant hyperthermia-susceptible human skeletal muscle. J. Biol. Chem. 2010, 285, 25645–25653. [Google Scholar] [CrossRef]
- Hopkins, P.M. Malignant hyperthermia: Pharmacology of triggering. Br. J. Anaesth. 2011, 107, 48–56. [Google Scholar] [CrossRef]
- Tong, J.; Oyamada, H.; Demaurex, N.; Grinstein, S.; McCarthy, T.V.; MacLennan, D.H. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J. Biol. Chem. 1997, 272, 26332–26339. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Daly, C.; Aboelsaod, E.M.; Gardner, L.; Hobson, S.J.; Riasat, K.; Shepherd, S.; Robinson, R.L.; Bilmen, J.G.; Gupta, P.K.; et al. Genetic epidemiology of malignant hyperthermia in the UK. Br. J. Anaesth. 2018, 121, 944–952. [Google Scholar] [CrossRef]
- Ke, T.; Gomez, C.R.; Mateus, H.E.; Castano, J.A.; Wang, Q.K. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a South American family. J. Hum. Genet. 2009, 54, 660–664. [Google Scholar] [CrossRef]
- Riazi, S.; Kraeva, N.; Hopkins, P.M. Malignant Hyperthermia in the Post-Genomics Era: New Perspectives on an Old Concept. Anesthesiology 2018, 128, 168–180. [Google Scholar] [CrossRef]
- Mungunsukh, O.; Deuster, P.; Muldoon, S.; O’Connor, F.; Sambuughin, N. Estimating prevalence of malignant hyperthermia susceptibility through population genomics data. Br. J. Anaesth. 2019, 123, e461–e463. [Google Scholar] [CrossRef]
- Jeong, S.K.; Kim, D.C.; Cho, Y.G.; Sunwoo, I.N.; Kim, D.S. A double mutation of the ryanodine receptor type 1 gene in a malignant hyperthermia family with multiminicore myopathy. J. Clin. Neurol. 2008, 4, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, D.C.; Lee, J.H.; Cho, Y.G.; Lee, H.S.; Choi, S.I.; Kim, D.S. Molecular genetic analysis of the ryanodine receptor gene (RYR1) in Korean malignant hyperthermia families. Korean J. Lab. Med. 2010, 30, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.Y.; Park, Y.E.; Shin, J.H.; Lee, C.H.; Jung, D.S.; Kim, D.S. Mild Clinical Features and Histopathologically Atypical Cores in Two Korean Families with Central Core Disease Harboring RYR1 Mutations at the C-terminal Region. J. Clin. Neurol. 2015, 11, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.N.; Park, H.J.; Lee, J.H.; Shin, H.Y.; Kim, S.H.; Kim, S.M.; Choi, Y.C. Clinical and Pathologic Findings of Korean Patients with RYR1-Related Congenital Myopathy. J. Clin. Neurol. 2018, 14, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Hoppe, K.; Hack, G.; Lehmann-Horn, F.; Jurkat-Rott, K.; Wearing, S.; Zullo, A.; Carsana, A.; Klingler, W. Hypermetabolism in B-lymphocytes from malignant hyperthermia susceptible individuals. Sci. Rep. 2016, 6, 33372. [Google Scholar] [CrossRef]
- Girard, T.; Cavagna, D.; Padovan, E.; Spagnoli, G.; Urwyler, A.; Zorzato, F.; Treves, S. B-lymphocytes from malignant hyperthermia-susceptible patients have an increased sensitivity to skeletal muscle ryanodine receptor activators. J. Biol. Chem. 2001, 276, 48077–48082. [Google Scholar] [CrossRef]
- McKinney, L.C.; Butler, T.; Mullen, S.P.; Klein, M.G. Characterization of ryanodine receptor-mediated calcium release in human B cells: Relevance to diagnostic testing for malignant hyperthermia. Anesthesiology 2006, 104, 1191–1201. [Google Scholar] [CrossRef]
- Tsien, R.Y.; Pozzan, T.; Rink, T.J. T-cell mitogens cause early changes in cytoplasmic free Ca2+ and membrane potential in lymphocytes. Nature 1982, 295, 68–71. [Google Scholar] [CrossRef]
- Berman, M.C. Characterisation of thapsigargin-releasable Ca(2+) from the Ca(2+)-ATPase of sarcoplasmic reticulum at limiting [Ca(2+)]. Biochim. Biophys. Acta 2000, 1509, 42–54. [Google Scholar] [CrossRef]
- Larach, M.G.; Localio, A.R.; Allen, G.C.; Denborough, M.A.; Ellis, F.R.; Gronert, G.A.; Kaplan, R.F.; Muldoon, S.M.; Nelson, T.E.; Ording, H.; et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology 1994, 80, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Dirksen, R.T.; Girard, T.; Gonsalves, S.G.; Hopkins, P.M.; Riazi, S.; Saddic, L.A.; Sambuughin, N.; Saxena, R.; Stowell, K.; et al. Variant curation expert panel recommendations for RYR1 pathogenicity classifications in malignant hyperthermia susceptibility. Genet. Med. 2021, 23, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.; Davis, M.; James, D.; Pollock, N.; Stowell, K. Malignant hyperthermia. Orphanet J. Rare. Dis. 2007, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.M.; Girard, T.; Dalay, S.; Jenkins, B.; Thacker, A.; Patteril, M.; McGrady, E. Malignant hyperthermia 2020: Guideline from the Association of Anaesthetists. Anaesthesia 2021, 76, 655–664. [Google Scholar] [CrossRef]
- Takeshima, H.; Nishimura, S.; Matsumoto, T.; Ishida, H.; Kangawa, K.; Minamino, N.; Matsuo, H.; Ueda, M.; Hanaoka, M.; Hirose, T.; et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature 1989, 339, 439–445. [Google Scholar] [CrossRef]
- Bevilacqua, J.A.; Monnier, N.; Bitoun, M.; Eymard, B.; Ferreiro, A.; Monges, S.; Lubieniecki, F.; Taratuto, A.L.; Laquerrière, A.; Claeys, K.G.; et al. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol. Appl. Neurobiol. 2011, 37, 271–284. [Google Scholar] [CrossRef]
- Witherspoon, J.W.; Meilleur, K.G. Review of RyR1 pathway and associated pathomechanisms. Acta Neuropathol. Commun. 2016, 4, 121. [Google Scholar] [CrossRef]
- Monnier, N.; Kozak-Ribbens, G.; Krivosic-Horber, R.; Nivoche, Y.; Qi, D.; Kraev, N.; Loke, J.; Sharma, P.; Tegazzin, V.; Figarella-Branger, D.; et al. Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Hum. Mutat. 2005, 26, 413–425. [Google Scholar] [CrossRef]
- Ferreiro, A.; Estournet, B.; Chateau, D.; Romero, N.B.; Laroche, C.; Odent, S.; Toutain, A.; Cabello, A.; Fontan, D.; dos Santos, H.G.; et al. Multi-minicore disease—Searching for boundaries: Phenotype analysis of 38 cases. Ann. Neurol. 2000, 48, 745–757. [Google Scholar] [CrossRef]
- Ferreiro, A.; Monnier, N.; Romero, N.B.; Leroy, J.P.; Bönnemann, C.; Haenggeli, C.A.; Straub, V.; Voss, W.D.; Nivoche, Y.; Jungbluth, H.; et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann. Neurol. 2002, 51, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, H.; Zhou, H.; Hartley, L.; Halliger-Keller, B.; Messina, S.; Longman, C.; Brockington, M.; Robb, S.A.; Straub, V.; Voit, T.; et al. Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology 2005, 65, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Jungbluth, H.; Sewry, C.A.; Feng, L.; Bertini, E.; Bushby, K.; Straub, V.; Roper, H.; Rose, M.R.; Brockington, M.; et al. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 2007, 130, 2024–2036. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.F.; Waddell, L.B.; Cooper, S.T.; Perry, M.; Smith, R.L.; Kornberg, A.J.; Muntoni, F.; Lillis, S.; Straub, V.; Bushby, K.; et al. Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum. Mutat. 2010, 31, E1544–E1550. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.M.; Rüffert, H.; Snoeck, M.M.; Girard, T.; Glahn, K.P.; Ellis, F.R.; Müller, C.R.; Urwyler, A. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br. J. Anaesth. 2015, 115, 531–539. [Google Scholar] [CrossRef]
- Larach, M.G.; North American Malignant Hyperthermia Group. Standardization of the caffeine halothane muscle contracture test. Anesth. Analg. 1989, 69, 511–515. [Google Scholar] [CrossRef]
- Allen, G.C.; Larach, M.G.; Kunselman, A.R. The sensitivity and specificity of the caffeine-halothane contracture test: A report from the North American Malignant Hyperthermia Registry. The North American Malignant Hyperthermia Registry of MHAUS. Anesthesiology 1998, 88, 579–588. [Google Scholar] [CrossRef]
- Riazi, S.; Kraeva, N.; Hopkins, P.M. Updated guide for the management of malignant hyperthermia. Can. J. Anaesth. 2018, 65, 709–721. [Google Scholar] [CrossRef]
- Sei, Y.; Gallagher, K.L.; Basile, A.S. Skeletal muscle type ryanodine receptor is involved in calcium signaling in human B lymphocytes. J. Biol. Chem. 1999, 274, 5995–6002. [Google Scholar] [CrossRef]
- Sei, Y.; Brandom, B.W.; Bina, S.; Hosoi, E.; Gallagher, K.L.; Wyre, H.W.; Pudimat, P.A.; Holman, S.J.; Venzon, D.J.; Daly, J.W.; et al. Patients with malignant hyperthermia demonstrate an altered calcium control mechanism in B lymphocytes. Anesthesiology 2002, 97, 1052–1058. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Le, N.T.; Nguyen, T.T.; Nguyen, H.H.; Nguyen, K.T.; Dinh, L.D.; Nguyen, T.B.; Do, A.T.; Nguyen, C.H.; Nguyen, T.H.; et al. Whole exome sequencing revealed a pathogenic variant in a gene related to malignant hyperthermia in a Vietnamese cardiac surgical patient: A case report. Ann. Med. Surg. 2019, 48, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.M.; Liao, M.H.; Chu, C.L.; Lin, Y.H.; Sun, W.Z.; Lai, L.P.; Chen, P.L. Next-generation sequencing and bioinformatics to identify genetic causes of malignant hyperthermia. J. Formos. Med. Assoc. 2020, 120, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, S.G.; Ng, D.; Johnston, J.J.; Teer, J.K.; Stenson, P.D.; Cooper, D.N.; Mullikin, J.C.; Biesecker, L.G. Using exome data to identify malignant hyperthermia susceptibility mutations. Anesthesiology 2013, 119, 1043–1053. [Google Scholar] [CrossRef]
- Nagele, P. Exome sequencing: One small step for malignant hyperthermia, one giant step for our specialty--why exome sequencing matters to all of us, not just the experts. Anesthesiology 2013, 119, 1006–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Gene | Genomic Position | Ref ID | Base Change /Codon Change | rsID | OMIM | gnomAD | Inheritance |
|---|---|---|---|---|---|---|---|
| RYR1 | chr19:38989754 | NM_000540.2 | c.6898T > C /p.Ser2300Pro | None | #145600, #117000 | 0 | Sporadic |
| GAA * | chr17:78081415 | NM_000152.5 | c.752C > T /p. Ser251Leu | rs200856561 | #232300 | 0.0003504 | Maternal |
| GAA * | chr17:78081424 | NM_000152.5 | c.761C > T /p. Ser254Leu | rs577915581 | #232300 | 0.0002071 | Maternal |
| GNE | chr9:36246117 | NM_001128227.3 | c.620A > T /p. Asp207Val | rs139425890 | #605820 | 0.00005173 | Paternal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, Y.J.; Park, J.; Kim, J.R.; Lee, S.Y.; Lee, J.; Cho, Y.G.; Kim, D.S. Gene Panel Sequencing Identifies a Novel RYR1 p.Ser2300Pro Variant as Candidate for Malignant Hyperthermia with Multi-Minicore Myopathy. Genes 2022, 13, 1726. https://doi.org/10.3390/genes13101726
Moon YJ, Park J, Kim JR, Lee SY, Lee J, Cho YG, Kim DS. Gene Panel Sequencing Identifies a Novel RYR1 p.Ser2300Pro Variant as Candidate for Malignant Hyperthermia with Multi-Minicore Myopathy. Genes. 2022; 13(10):1726. https://doi.org/10.3390/genes13101726
Chicago/Turabian StyleMoon, Young Jae, Joonhong Park, Jung Ryul Kim, Seung Yeob Lee, Jaehyeon Lee, Yong Gon Cho, and Dal Sik Kim. 2022. "Gene Panel Sequencing Identifies a Novel RYR1 p.Ser2300Pro Variant as Candidate for Malignant Hyperthermia with Multi-Minicore Myopathy" Genes 13, no. 10: 1726. https://doi.org/10.3390/genes13101726