
The Genetic Diversity and the Divergence Time in Extant Primitive Mayfly, Siphluriscus chinensis Ulmer, 1920 Using the Mitochondrial Genome
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Morphological Identification
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Gene Annotation and Sequence Analyses
2.4. Phylogenetic Analyses
2.5. Divergence Time Estimation
3. Results
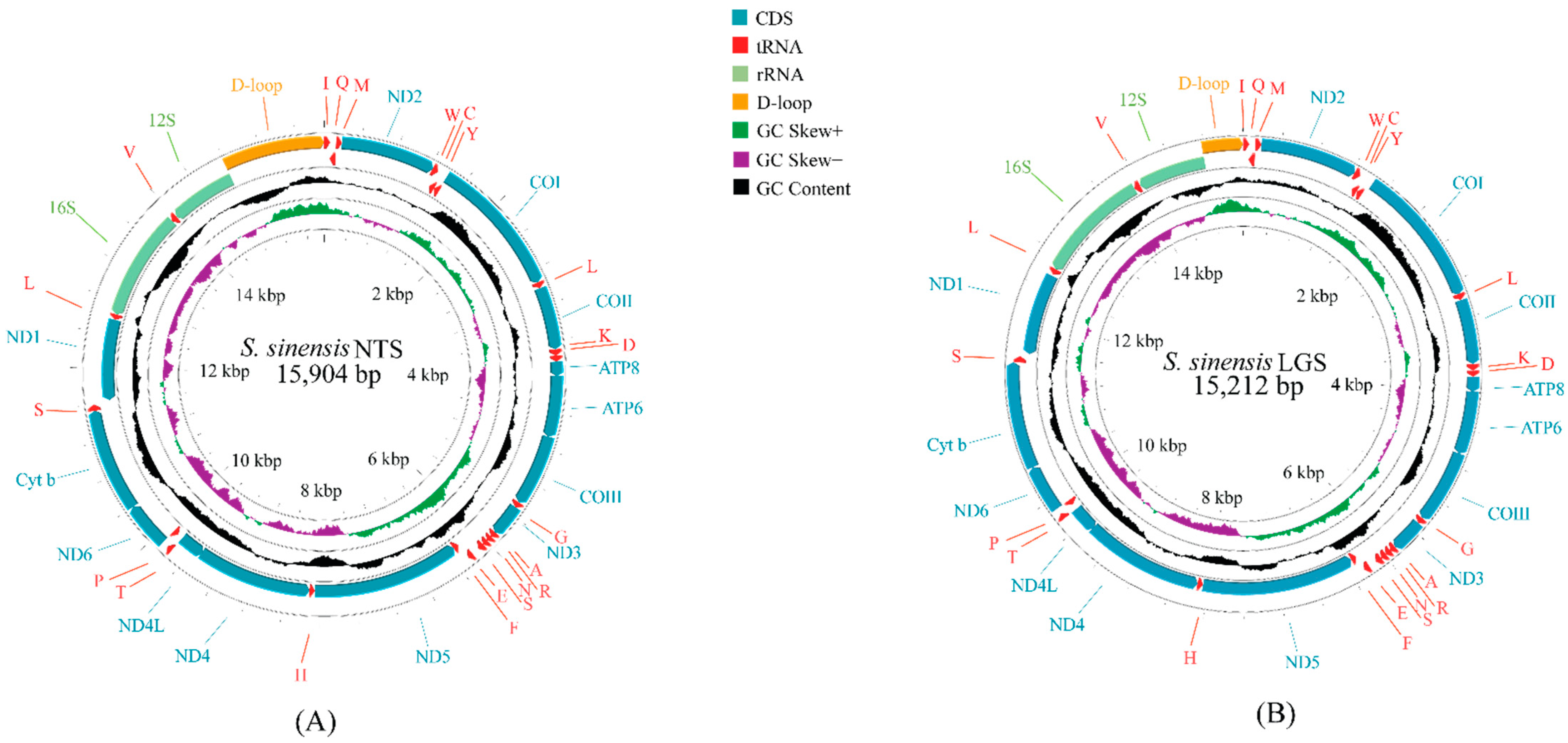
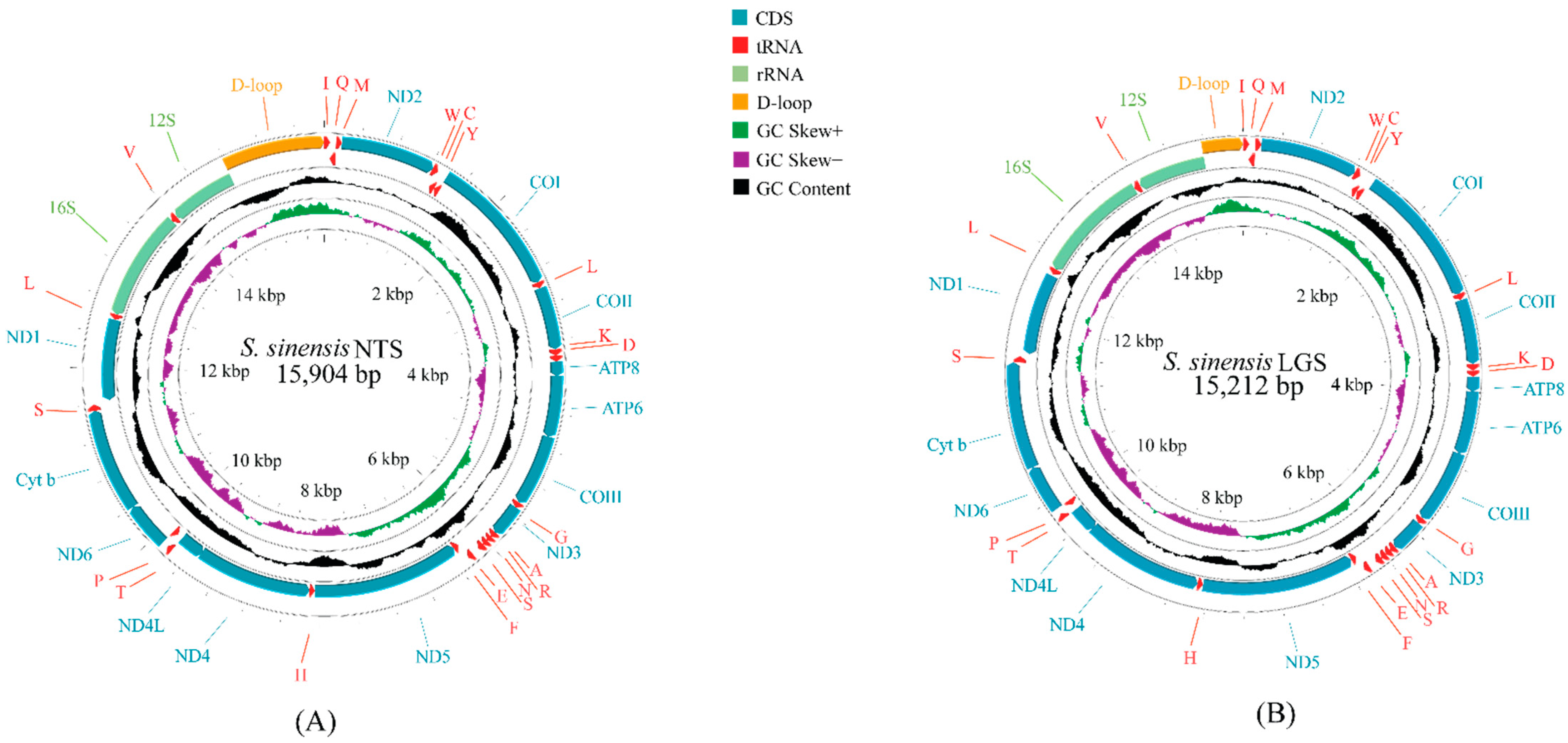
3.1. Mitochondrial Genome Organization
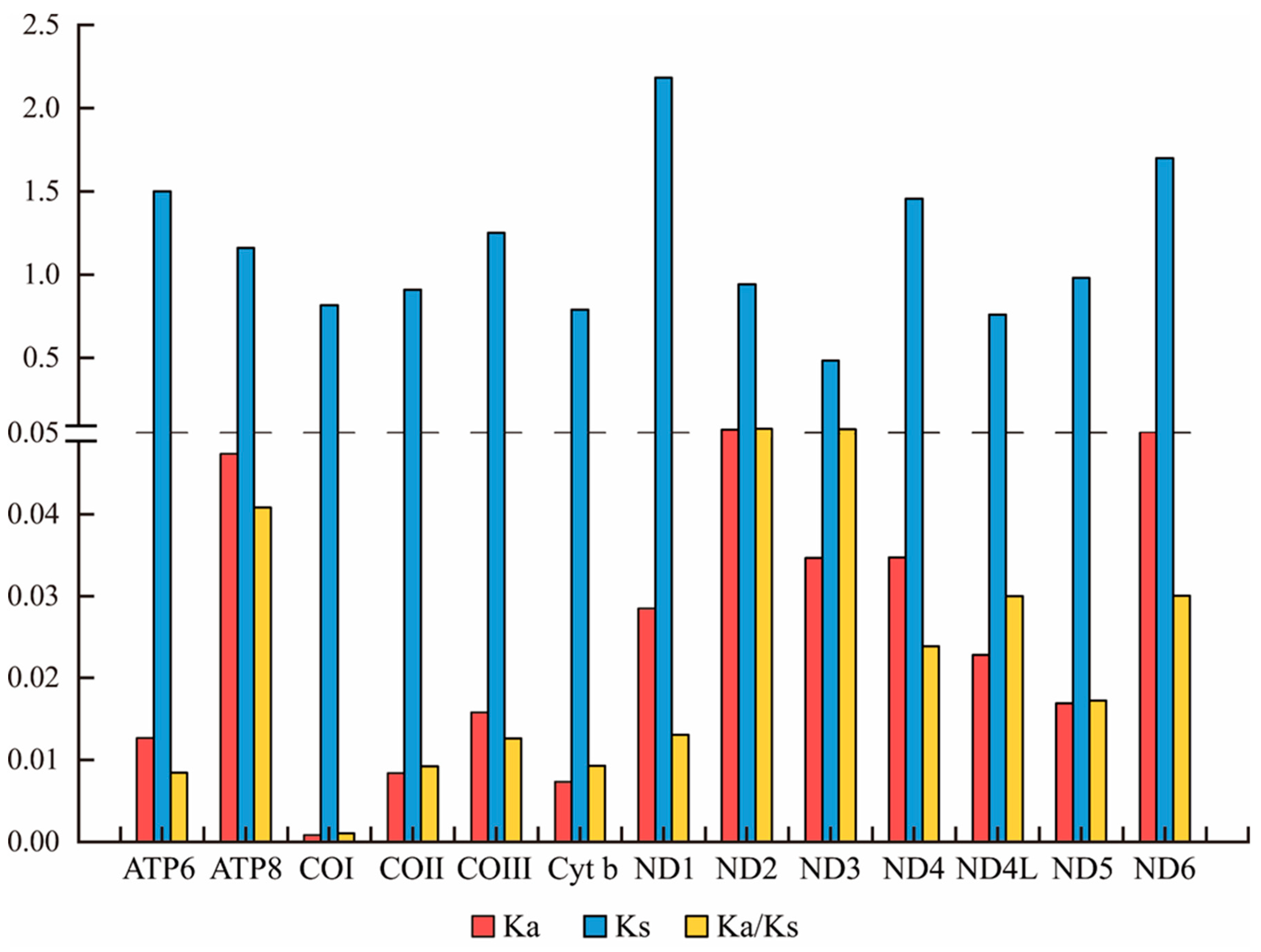
3.2. Calculation of Genetic Distance
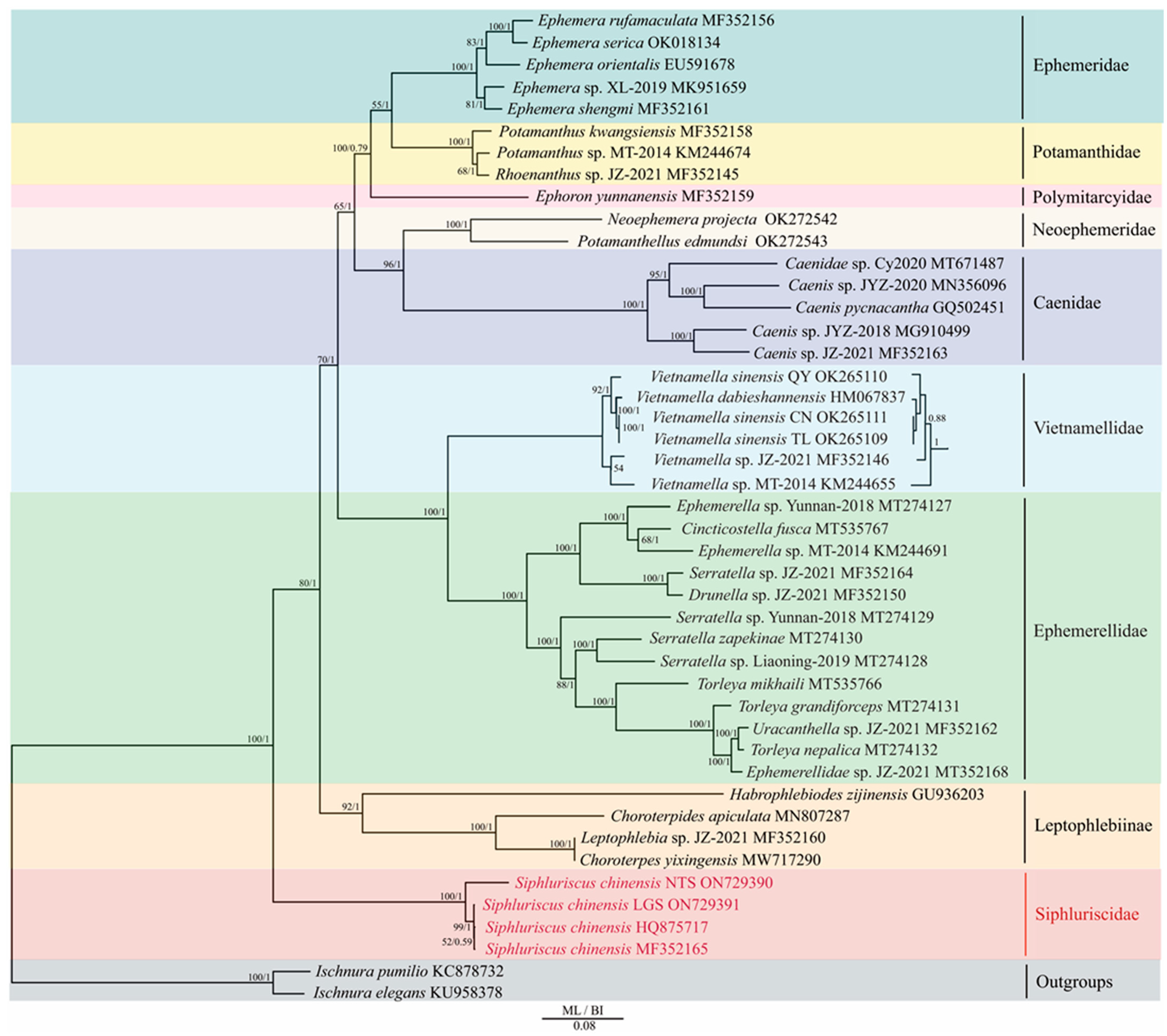
3.3. Phylogenetic Analyses
3.4. Divergence Time Estimation
4. Discussion
4.1. Composition Differences in Mitochondrial Genomes
4.2. Phylogenetic Analyses and Identification of Cryptic Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ogden, T.H.; Gattolliat, J.L.; Sartori, M.; Staniczek, A.H.; Soldán, T.; Whiting, M.F. Towards a new paradigm in mayfly phylogeny (Ephemeroptera): Combined analysis of morphological and molecular data. Syst. Entomol. 2009, 34, 616–634. [Google Scholar] [CrossRef]
- Barber-James, H.M.; Gattolliat, J.L.; Sartori, M.; Hubbard, M. Global diversity of mayflies (Ephemeroptera, Insecta) in freshwater. Hydrobiologia 2008, 595, 339–350. [Google Scholar] [CrossRef]
- Bauernfeind, E.; Soldán, T. The mayflies of Europe (Ephemeroptera). Apollo Books Ollerup Den. 2012, 61, 190–191. [Google Scholar]
- Jacobus, L.M.; Macadam, C.R.; Sartori, M. Mayflies (Ephemeroptera) and their contributions to ecosystem services. Insects 2019, 10, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Qin, J.C.; Zhou, C. The phylogeny of Ephemeroptera in Pterygota revealed by the mitochondrial genome of Siphluriscus chinensis (Hexapoda: Insecta). Gene 2014, 545, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Li, X.; Yin, X.; Li, X.; Yin, J.; Pan, P. The mitochondrial genomes of palaeopteran insects and insights into the early insect relationships. Sci. Rep. 2019, 9, 17765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.N.; Yu, P.P.; Zhang, L.P.; Storey, K.B.; Zhang, J.Y. Increasing 28 mitogenomes of Ephemeroptera, Odonata and Plecoptera support the Chiastomyaria hypothesis with three different outgroup combinations. PeerJ 2021, 9, e11402. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.Y.; Gao, Y.J.; Zhang, L.P.; Yu, D.N.; Zhang, J.Y. The mitochondrial genome of Caenis sp. (Ephemeroptera: Caenidae) and the phylogeny of Ephemeroptera in Pterygota. Mitochondrial DNA B 2018, 3, 577–579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, R.; Zhou, C.F. Complete mitochondrial genomes of Epeorus carinatus and E. dayongensis (Ephemeroptera: Heptageniidae): Genomic comparison and phylogenetic inference. Gene 2021, 777, 145467–145475. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Ma, Z.; Zhou, C. First complete mitogenomes of three mayflies in the genus Afronurus (Ephemeroptera: Heptageniidae) and their implications for phylogenetic reconstruction. Biologia 2021, 76, 2291–2302. [Google Scholar] [CrossRef]
- Ulmer, G. Neue Ephemeropteren. Arch. Naturg. 1920, 85, 1–80. [Google Scholar]
- Demoulin, G. Brève note sur la position systématique de Siphluriscus chinensis Ulmer (1920)(Ephemeroptera). Bull. Inst. R. Sci. Nat. Belg. 1955, 31, 1–2. [Google Scholar]
- Edmunds, G.F.; Koss, R.W. Review of the Acanthametropodinae with a description of a new genus (Ephemeroptera: Siphlonuridae). Pan Pac. Entomol. 1972, 48, 136–144. [Google Scholar]
- Demoulin, G. Remarques critiques sur les Acanthametropodinae et sur certaines formes affines (Ephemeroptera: Siphlonuridae). Bull. Inst. R. Sci. Nat. Belg. Entomol. 1974, 50, 1–5. [Google Scholar]
- McCafferty, W.; Wang, T. Relationships of the genera Acanthametropus, Analetris, and Siphluriscus, and re-evaluation of their higher classification (Ephemeroptera: Pisciforma). Great Lakes Entomol. 1994, 27, 209–215. [Google Scholar]
- Zhou, C.F.; Peters, J.G. The nymph of Siphluriscus chinensis and additional imaginal description: A living mayfly with Jurassic origins (Siphluriscidae new family: Ephemeroptera). Fla. Entomol. 2003, 86, 345–352. [Google Scholar] [CrossRef]
- Van Vinh Nguyen, V.H.N.; Bae, Y.J. A Rare Mayfly Siphluriscus chinensis Ulmer (Ephemeroptera: Siphluriscidae) from Vietnam. Entomol. Res. Bull. 2015, 31, 56–57. [Google Scholar]
- Edmunds, G.F. Biogeography and evolution of Ephemeroptera. Annu. Rev. Entomol. 1972, 17, 21–42. [Google Scholar] [CrossRef]
- Wang, T.Q.; McCafferty, W.P. Heptageniidae (Ephemeroptera) of the World. Part I: Phylogenetic higher classification. Trans. Am. Entomol. Soc. 2004, 130, 11–45. [Google Scholar]
- O’Donnell, B.C.; Jockusch, E.L. Phylogenetic relationships of leptophlebiid mayflies as inferred by histone H3 and 28S ribosomal DNA. Syst. Entomol. 2008, 33, 651–667. [Google Scholar] [CrossRef]
- Kluge, N.J. Higher system of Atalophlebiinae (Leptophlebiidae) with description of three new species of Terpides s. l. from Peruvian Amazonia. Russ. Entomol. J. 2009, 18, 243–256. [Google Scholar]
- Zhang, J.Y.; Zhang, L.P.; Yu, D.; Zheng, R.Q. Complete mitochondrial genomes of Nanorana taihangnica and N. yunnanensis (Anura: Dicroglossidae) with novel gene arrangements and phylogenetic relationship of Dicroglossidae. BMC Evol. Biol. 2018, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.D.; Jia, Y.Y.; Cao, S.S.; Zhang, Z.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Six complete mitochondrial genomes of mayflies from three genera of Ephemerellidae (Insecta: Ephemeroptera) with inversion and translocation of trnI rearrangement and their phylogenetic relationships. PeerJ 2020, 8, e9740. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.Y.; Shen, S.Q.; Zhang, Z.Y.; Xu, X.D.; Yu, D.N.; Zhang, J.Y. Comparative mitogenomes of two Coreamachilis species (Microcoryphia: Machilidae) along with phylogenetic analyses of Microcoryphia. Insects 2021, 12, 795. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol. Evol. 2006, 21, 439–446. [Google Scholar] [CrossRef]
- Gao, X.Y.; Cai, Y.Y.; Yu, D.N.; Zhang, J.Y. Characteristics of the complete mitochondrial genome of Suhpalacsa longialata (Neuroptera, Ascalaphidae) and its phylogenetic implications. PeerJ 2018, 6, e5914. [Google Scholar] [CrossRef] [Green Version]
- Ayivi, S.P.G.; Tong, Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The mitochondrial genomes of 18 new Pleurosticti (Coleoptera: Scarabaeidae) exhibit a novel trnQ-NCR-trnI-trnM gene rearrangement and clarify phylogenetic relationships of subfamilies within Scarabaeidae. Insects 2021, 12, 1025. [Google Scholar] [CrossRef]
- Xu, K.K.; Chen, Q.P.; Ayivi, S.P.G.; Guan, J.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Three complete mitochondrial genomes of Orestes guangxiensis, Peruphasma schultei, and Phryganistria guangxiensis (Insecta: Phasmatodea) and their phylogeny. Insects 2021, 12, 779. [Google Scholar] [CrossRef]
- Blouin, M.S. Molecular prospecting for cryptic species of nematodes: Mitochondrial DNA versus internal transcribed spacer. Int. J. Parasitol. 2002, 32, 527–531. [Google Scholar] [CrossRef]
- Yang, Y.M.; Zhang, L.H.; Lin, Y.J.; Zheng, Y.M.; Jin, W.T.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The genetic diversity in Thereuonema tuberculata (Wood, 1862) (Scutigeromorpha: Scutigeridae) and the phylogenetic relationship of Scutigeromorpha using the mitochondrial genome. Insects 2022, 13, 620. [Google Scholar] [CrossRef] [PubMed]
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K.L.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2007, 22, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Blair, D. Relative merits of nuclear ribosomal internal transcribed spacers and mitochondrial CO1 and ND1 genes for distinguishing among Echinostoma species (Trematoda). Parasitology 1998, 116, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Haruyama, N.; Naka, H.; Mochizuki, A.; Nomura, M. Mitochondrial phylogeny of cryptic species of the lacewing Chrysoperla nipponensis (Neuroptera: Chrysopidae) in Japan. Ann. Entomol. Soc. Am. 2008, 101, 971–977. [Google Scholar] [CrossRef]
- Feng, S.; Yang, Q.; Li, H.; Song, F.; Stejskal, V.; Opit, G.P.; Ca, I.W.; Li, Z.; Shao, R. The highly divergent mitochondrial genomes indicate that the booklouse, Liposcelis bostrychophila (Psocoptera: Liposcelididae) is a cryptic species. G3-Genes Genomes Genet. 2018, 8, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.N.; Zhang, J.Y.; Peng, L.; Zheng, R.Q.; Shao, C. Do cryptic species exist in Hoplobatrachus rugulosus? An examination using four nuclear genes, the Cyt b gene and the complete mt genome. PLoS ONE 2015, 10, e0124825. [Google Scholar] [CrossRef]
- Justine, J.L.; Gastineau, R.; Gros, P.; Gey, D.; Ruzzier, E.; Charles, L.; Winsor, L. Hammerhead flatworms (Platyhelminthes, Geoplanidae, Bipaliinae): Mitochondrial genomes and description of two new species from France, Italy, and Mayotte. PeerJ 2022, 10, e12725. [Google Scholar] [CrossRef]
- Souto, P.M.; da Silveira, L.F.L.; Takiya, D.M.; Salles, F.F. Cryptic diversity in the mayfly Leptohyphodes inanis (Pictet)(Ephemeroptera: Leptohyphidae) across water basins in Southeastern Brazil. Syst. Biodivers. 2021, 19, 797–817. [Google Scholar] [CrossRef]
- Ståhls, G.; Savolainen, E. MtDNA COI barcodes reveal cryptic diversity in the Baetis vernus group (Ephemeroptera, Baetidae). Mol. Phylogenet. Evol. 2008, 46, 82–87. [Google Scholar] [CrossRef]
- Golding, M. Real World Adobe Illustrator CS4; Peachpit Press: Berkeley, CA, USA, 2008. [Google Scholar]
- Zhang, J.Y.; Zhou, C.F.; Gai, Y.H.; Song, D.X.; Zhou, K.Y. The complete mitochondrial genome of Parafronurus youi (Insecta: Ephemeroptera) and phylogenetic position of the Ephemeroptera. Gene 2008, 424, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, S. Primer premier 5. Biotech. Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. In Bioinformatics Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2000; pp. 71–91. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Xu, X.D.; Jia, Y.Y.; Dai, X.Y.; Ma, J.L.; Yu, D.N. The mitochondrial genome of Caenis sp. (Ephemeroptera: Caenidae) from Fujian and the phylogeny of Caenidae within Ephemeroptera. Mitochondrial DNA B 2019, 5, 192–193. [Google Scholar] [CrossRef] [Green Version]
- Macher, J.N.; Drakou, K.; Papatheodoulou, A.; Van Der Hoorn, B.; Vasquez, M. The mitochondrial genomes of 11 aquatic macroinvertebrate species from Cyprus. Metabarcoding Metagenom. 2020, 4, e58259. [Google Scholar] [CrossRef]
- Li, R.; Ma, Z.X.; Zhou, C.F. The first two complete mitochondrial genomes of Neoephemeridae (Ephemeroptera): Comparative analysis and phylogenetic implication for Furcatergalia. Genes 2021, 12, 1875. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.M.; Hong, M.Y.; Kim, M.I.; Kim, M.J.; Park, H.C.; Kim, K.Y.; Lee, I.H.; Bae, C.H.; Jin, B.R.; Kim, I. The complete mitogenome sequences of the palaeopteran insects Ephemera orientalis (Ephemeroptera: Ephemeridae) and Davidius lunatus (Odonata: Gomphidae). Genome 2009, 52, 810–817. [Google Scholar] [CrossRef]
- Tang, M.; Tan, M.; Meng, G.; Yang, S.; Su, X.; Liu, S.; Song, W.; Li, Y.; Wu, Q.; Zhang, A. Multiplex sequencing of pooled mitochondrial genomes—A crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 2014, 42, e166. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhang, W.; Ma, Z.X.; Zhou, C.F. Novel gene rearrangement pattern in the mitochondrial genomes of Torleya mikhaili and Cincticostella fusca (Ephemeroptera: Ephemerellidae). Int. J. Biol. Macromol. 2020, 165, 3106–3114. [Google Scholar] [CrossRef]
- Tong, Y.; Wu, L.; Ayivi, S.P.G.; Storey, K.B.; Ma, Y.; Yu, D.N.; Zhang, J.Y. Cryptic species exist in Vietnamella sinensis Hsu, 1936 (Insecta: Ephemeroptera) from studies of complete mitochondrial genomes. Insects 2022, 13, 412. [Google Scholar] [CrossRef]
- Cao, S.S.; Xu, X.D.; Jia, Y.Y.; Guan, J.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The complete mitochondrial genome of Choroterpides apiculata (Ephemeroptera: Leptophlebiidae) and its phylogenetic relationships. Mitochondrial DNA B 2020, 5, 1159–1160. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.Y.; Zhang, Z.Y.; Cao, Y.R.; Xu, X.D.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The complete mitochondrial genome of Choroterpes (Euthralus) yixingensis (Ephemeroptera: Leptophlebiidae) and its mitochondrial protein-coding gene expression under imidacloprid stress. Gene 2021, 800, 145833. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, B.; Jiang, J.; Tong, X. The complete mitochondrial genome of Ephemera serica (Ephemeroptera: Ephemeridae) and phylogenetic analysis. Mitochondrial DNA B 2022, 7, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Carballa, M.O.; Thompson, D.J.; Cordero-Rivera, A.; Watts, P.C. Next Generation Sequencing Yields the Complete Mitochondrial Genome of the Scarce Blue-Tailed Damselfly, Ischnura Pumilio; Taylor & Francis: Abingdon, UK, 2014. [Google Scholar]
- Feindt, W.; Herzog, R.; Osigus, H.-J.; Schierwater, B.; Hadrys, H. Short read sequencing assembly revealed the complete mitochondrial genome of Ischnura elegans Vander Linden, 1820 (Odonata: Zygoptera). Mitochondrial DNA Part B 2016, 1, 574–576. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE–visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinf. 2014, 15, 294. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A. Figtree Version 1.4.0. 2012. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 June 2022).
- Kishino, H.; Thorne, J.L.; Bruno, W.J. Performance of a divergence time estimation method under a probabilistic model of rate evolution. Mol. Biol. Evol. 2001, 18, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, P.C.; Benton, M.J. Rocks and clocks: Calibrating the Tree of Life using fossils and molecules. Trends Ecol. Evol. 2007, 22, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Cobbett, A.; Wilkinson, M.; Wills, M.A. Fossils impact as hard as living taxa in parsimony analyses of morphology. Syst. Biol. 2007, 56, 753–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.Y.; Phillips, M.J. Accounting for calibration uncertainty in phylogenetic estimation of evolutionary divergence times. Syst. Biol. 2009, 58, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyron, R.A. Divergence time estimation using fossils as terminal taxa and the origins of Lissamphibia. Syst. Biol. 2011, 60, 466–481. [Google Scholar] [CrossRef] [Green Version]
- Staniczek, A.H.; Godunko, R.J.; Krzeminski, W. A new fossil mayfly species of the genus Borinquena Traver, 1938 (Insecta: Ephemeroptera: Leptophlebiidae: Atalophlebiinae) from Miocene Dominican amber. Ann. Zool. 2017, 67, 113–119. [Google Scholar] [CrossRef]
- Zhang, W.T.; Shih, C.K.; Shih, Y.H.; Ren, D. A new macrolepidopteran moth (Insecta, Lepidoptera, Geometridae) in Miocene Dominican amber. Zookeys 2020, 965, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Staniczek, A.H.; Godunko, R.J.; Kluge, N.J. Fossil record of the mayfly family Ephemerellidae (Insecta: Ephemeroptera), with description of new species and first report of Ephemerellinae from Baltic amber. J. Syst. Palaeontol. 2018, 16, 1319–1335. [Google Scholar] [CrossRef]
- Godunko, R.J.; Martynov, A.V.; Staniczek, A.H. First fossil record of the mayfly family Vietnamellidae (Insecta, Ephemeroptera) from Burmese amber confirms its Oriental origin and gives new insights into its evolution. Zookeys 2021, 1036, 99–120. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.L.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zhu, K.; Liu, Y.; Zhang, H.; Gong, L.; Jiang, L.; Liu, L.; Lü, Z.; Liu, B. Novel gene rearrangement in the mitochondrial genome of Muraenesox cinereus and the phylogenetic relationship of Anguilliformes. Sci. Rep. 2021, 11, 2411. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.P.; Tsai, M.H.; Kroh, A.; Trautman, A.; Machado, D.J.; Chang, L.Y.; Reid, R.; Lin, K.T.; Bronstein, O.; Lee, S.J. The first complete mitochondrial genome of the sand dollar Sinaechinocyamus mai (Echinoidea: Clypeasteroida). Genomics 2020, 112, 1686–1693. [Google Scholar] [CrossRef]
- Prabhu, V.R.; Singha, H.S.; Kumar, R.G.; Gopalakrishnan, A.; Nagarajan, M. Characterization of the complete mitochondrial genome of Barilius malabaricus and its phylogenetic implications. Genomics 2020, 112, 2154–2163. [Google Scholar] [CrossRef]
- Xu, T.J.; Cheng, Y.Z.; Sun, Y.N.; Shi, G.; Wang, R.X. The complete mitochondrial genome of bighead croaker, Collichthys niveatus (Perciformes, Sciaenidae): Structure of control region and phylogenetic considerations. Mol. Biol. Rep. 2011, 38, 4673–4685. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.G.; Krieger, M.; Shoemaker, D.W.; Vargo, E.L.; Keller, L. Hierarchical analysis of genetic structure in native fire ant populations: Results from three classes of molecular markers. Genetics 1997, 147, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.C.; Ormerod, S.J.; Bruford, M.W. Molecular systematics and phylogeography of the cryptic species complex Baetis rhodani (Ephemeroptera, Baetidae). Mol. Phylogenetics Evol. 2006, 40, 370–382. [Google Scholar] [CrossRef] [PubMed]
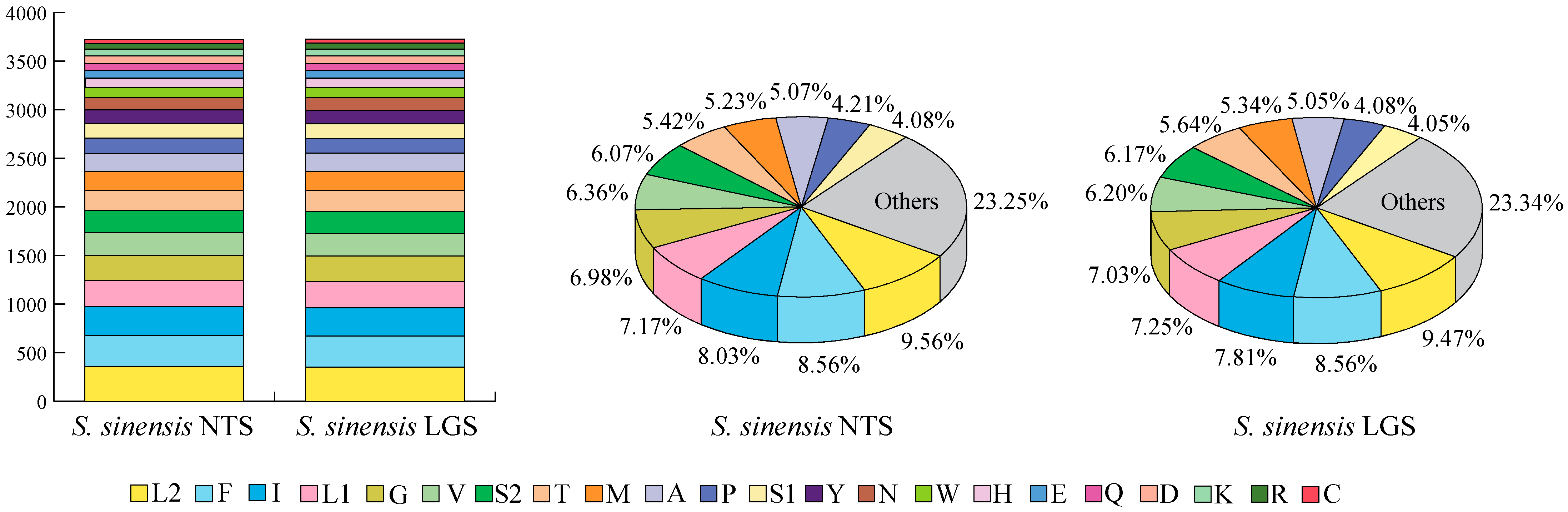



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Strand | S. chinensis NTS | S. chinensis LGS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | AT% | AT Skew | GC Skew | Length (bp) | AT% | AT Skew | GC Skew | ||
| Whole genome | 15,904 | 65.8 | 0.025 | −0.166 | 15,212 | 66.6 | 0.021 | −0.159 | |
| PCGs | + | 6876 | 63.6 | −0.136 | −0.178 | 6879 | 64.7 | −0.145 | −0.159 |
| - | 4329 | 67.7 | −0.256 | 0.197 | 4329 | 68.2 | −0.244 | 0.190 | |
| tRNAs | + | 908 | 66.2 | −0.028 | 0.121 | 907 | 66.1 | −0.010 | 0.111 |
| - | 523 | 70.0 | −0.055 | 0.312 | 523 | 70.4 | −0.049 | 0.290 | |
| rRNAs | - | 2074 | 68.2 | −0.074 | 0.236 | 2064 | 69.4 | −0.086 | 0.281 |
| Species | GenBank No. | 1 | 2 | 3 | |
|---|---|---|---|---|---|
| 1 | S. chinensis | HQ875717 | |||
| 2 | S. chinensis | MF352165 | 0.003 | ||
| 3 | S. chinensis LGS | ON729390 | 0.003 | 0.002 | |
| 4 | S. chinensis NTS | ON729391 | 0.123 | 0.122 | 0.123 |
| Nodes/Clades | Mean Divergence Time (Mya) | 95% HPD Range (Mya) |
|---|---|---|
| Ephemeridae & Potamanthidae | 86.07 | 56.35~117.99 |
| (Ephemeridae + Potamanthidae) & Polymitarcyidae | 108.06 | 78.07~134.65 |
| Neoephemeridae & Caenidae | 106.45 | 83.84~129.98 |
| (Neoephemeridae + Caenida) & (Ephemeridae + Potamanthidae+ Polymitarcyidae) | 126.79 | 105.02~148.76 |
| Vietnamellidae & Ephemerellidae | 98.51 | 98.00~99.00 |
| (Vietnamellidae + Ephemerellidae) & ((Neoephemeridae + Caenida) + ((Ephemeridae + Potamanthidae) + Polymitarcyidae)) | 138.10 | 119.64~159.06 |
| Leptophlebiinae & ((Vietnamellidae + Ephemerellidae) + ((Neoephemeridae + Caenida) + ((Ephemeridae + Potamanthidae) + Polymitarcyidae))) | 149.45 | 129.50~171.22 |
| (Leptophlebiinae + ((Vietnamellidae + Ephemerellidae) + ((Neoephemeridae + Caenida) + ((Ephemeridae + Potamanthidae) + Polymitarcyidae)))) & Siphluriscidae | 174.43 | 163.38~197.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, Y.; Shen, C.-Y.; Zhao, Y.-Y.; Lin, Y.-J.; Wu, L.; Storey, K.B.; Yu, D.-N.; Zhang, J.-Y. The Genetic Diversity and the Divergence Time in Extant Primitive Mayfly, Siphluriscus chinensis Ulmer, 1920 Using the Mitochondrial Genome. Genes 2022, 13, 1780. https://doi.org/10.3390/genes13101780
Tong Y, Shen C-Y, Zhao Y-Y, Lin Y-J, Wu L, Storey KB, Yu D-N, Zhang J-Y. The Genetic Diversity and the Divergence Time in Extant Primitive Mayfly, Siphluriscus chinensis Ulmer, 1920 Using the Mitochondrial Genome. Genes. 2022; 13(10):1780. https://doi.org/10.3390/genes13101780
Chicago/Turabian StyleTong, Yao, Chen-Yang Shen, Yu-Yang Zhao, Yi-Jie Lin, Lian Wu, Kenneth B. Storey, Dan-Na Yu, and Jia-Yong Zhang. 2022. "The Genetic Diversity and the Divergence Time in Extant Primitive Mayfly, Siphluriscus chinensis Ulmer, 1920 Using the Mitochondrial Genome" Genes 13, no. 10: 1780. https://doi.org/10.3390/genes13101780