
Supervised Machine Learning Enables Geospatial Microbial Provenance
 , ,
, ,
Abstract
:1. Introduction
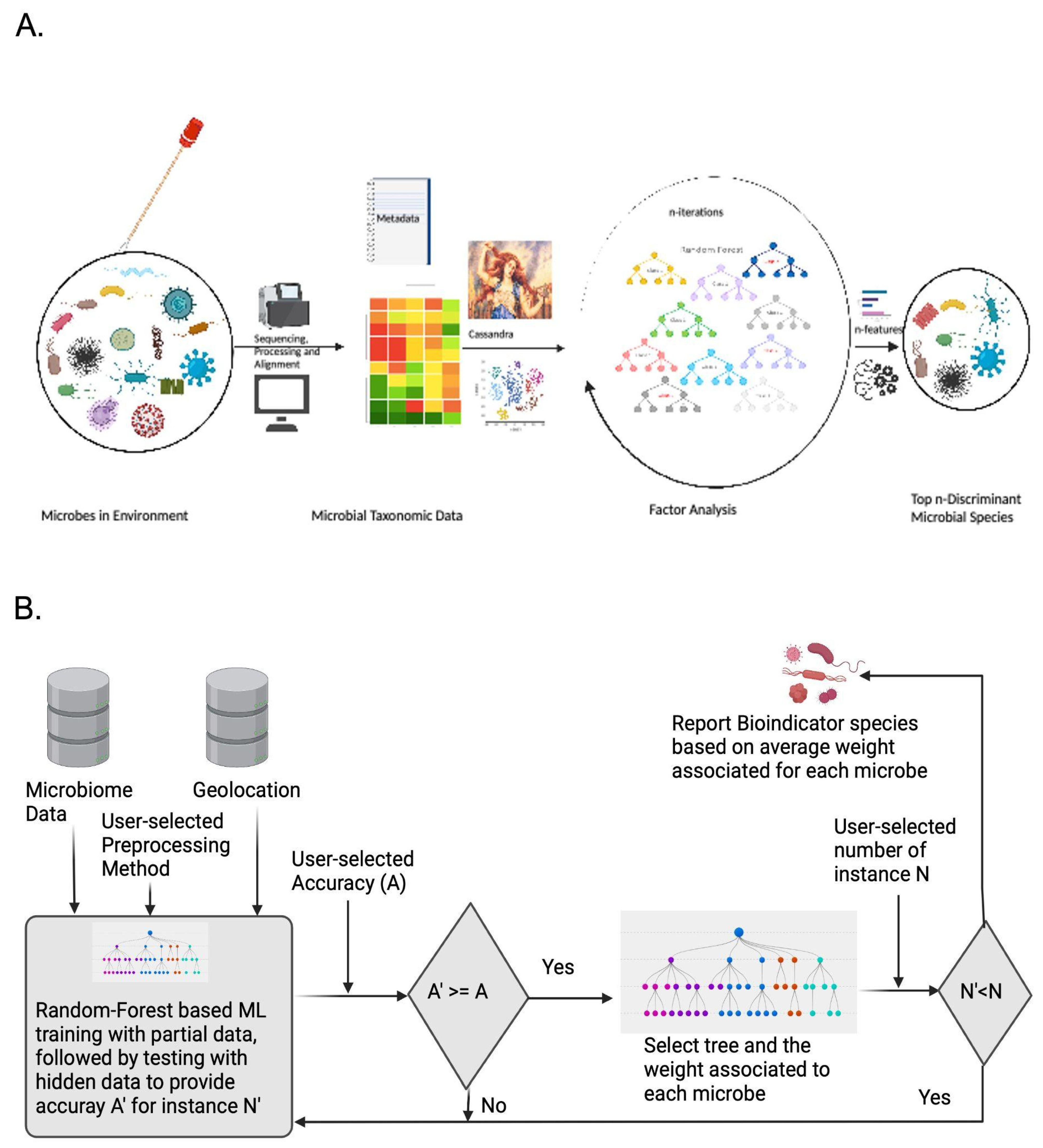
2. Methods
2.1. Dataset Details
2.2. Data Preprocessing and Adopted Classification Methodologies
- binary: Binarizing to 0,1 based on a threshold value (default = 0.0001).
- clr: Transformation function for compositional data based on Aitchison geometry to the real space.
- multiplicative replacement: Transformation function for compositional data uses the multiplicative replacement strategy for replacing zeros such that compositions still add up to 1.
- raw: No preprocessing.
- standard scalar: Standardizing the data by removing the mean and scaling to unit variance.
- total-sum: Converts all the samples to relative abundance.
- (1)
- Logistic Regression, which classifies estimating the probability of an event occurring based on a priori knowledge.
- (2)
- Linear Discriminant Analysis (LDA), a generalization of Fisher’s linear discriminant for separating multiple classes based on linear combination of features.
- (3)
- Ensemble Methods, which are an amalgamation of multiple models to produce an optimal classification (Random Forest Classifier, Extra Tree, AdaBoost).
- (4)
- Tree-based Classifiers including Decision Tree that use branching methods to evaluate the outcomes for classification.
- (5)
- K-nearest neighbor (kNN), a Supervised Learning method which classifies it by a vote of K nearest training objects as determined by some distance metric.
- (6)
- Bayesian Classifiers, including Gaussian NBC which update their posterior probability after ingesting new kinds of data.
- (7)
- Support Vector Machines (SVM), which separates classes by using a maximal margin hyperplane based on nonlinear decision boundary (Support Vector Classifier and Linear Support Vector Classifier (lSCV)).
2.3. Addressing Overfitting by Cross-Validation
2.4. Cassandra Predicts Bioindicator Species Providing Explainability and Interpretability of Datasets
3. Results
3.1. Microbial Fingerprints can Be Observed from the MetaSUB Dataset Both at the City and Continent Levels
3.2. Adding Interpretability and Explainability to Machine-Learned Microbial Fingerprints by Characterizing Bioindicator Microbes with Cassandra
3.3. Feature Interpolation for Microbial Forensics can Be Achieved Using Microbial Data
3.4. Modeling the Microbial Fingerprint of the Tara Oceans Dataset
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DNA | Deoxyribonucleic acid |
| ITS | Internal Transcribed Spacer |
| kNN | k-Nearest Neighbors |
| LDA | Linear Discriminant Analysis |
| LOGO | Leave one Group Out |
| lSVC | Linear Support Vector Classifier |
| MetaSUB | The Metagenomics and Metadesign of the Subways and Urban Biomes |
| MF | Microbial Forensics |
| ML | Machine Learning |
| NBC | Naive Bayes Classifier |
| OTU | Operational Taxonomic Unit |
| RF | Random Forest (Classifier) |
| rDNA | Recombinant DNA |
| rRNA | Ribosomal Ribonucleic Acid |
| SML | Supervised Machine Learning |
| SVM | Support Vector Machine |
| WGS | Whole genome sequencing |
References
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth Microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesant, S.; Not, F.; Picheral, M.; Kandels-Lewis, S.; Le Bescot, N.; Gorsky, G.; Iudicone, D.; Karsenti, E.; Speich, S.; Troublé, R.; et al. Open science resources for the discovery and analysis of Tara Oceans data. Sci. Data 2015, 2, 150023. [Google Scholar] [CrossRef] [Green Version]
- Danko, D.; Bezdan, D.; Afshin, E.E.; Ahsanuddin, S.; Bhattacharya, C.; Butler, D.J.; Chng, K.R.; Donnellan, D.; Hecht, J.; Jackson, K.; et al. A global metagenomic map of urban microbiomes and antimicrobial resistance. Cell 2021, 184, 3376–3393. [Google Scholar] [CrossRef] [PubMed]
- Ryon, K.A.; Tierney, B.T.; Frolova, A.; Kahles, A.; Desnues, C.; Ouzounis, C.; Gibas, C.; Bezdan, D.; Deng, Y.; He, D.; et al. A history of the MetaSUB consortium: Tracking urban microbes around the globe. iScience 2022. [CrossRef]
- Tighe, S.; Afshinnekoo, E.; Rock, T.M.; McGrath, K.; Alexander, N.; McIntyre, A.; Ahsanuddin, S.; Bezdan, D.; Green, S.J.; Joye, S.; et al. Genomic methods and microbiological technologies for profiling novel and extreme environments for the Extreme Microbiome Project (XMP). J. Biomol. Tech. JBT 2017, 28, 31. [Google Scholar] [CrossRef]
- Sierra, M.; Ryon, K.; Tierney, B.; Foox, J.; Bhattacharya, C.; Afshin, E.; Butler, D.; Green, S.; Thomas, K.; Ramsdell, J.; et al. Cross-kingdom metagenomic profiling of Lake Hillier reveals pigment-rich polyextremophiles and wide-ranging metabolic adaptations. BioRxiv 2022. [CrossRef]
- Tighe, S.; Baldwin, D.; Green, S.; Reyero, N. Next-generation sequencing and the extreme microbiome project (XMP). Next Gener. Seq. Appl. 2015, 2, 2. [Google Scholar] [CrossRef]
- Elhaik, E.; Ahsanuddin, S.; Robinson, J.M.; Foster, E.M.; Mason, C.E. The impact of cross-kingdom molecular forensics on genetic privacy. Microbiome 2021, 9, 114. [Google Scholar] [CrossRef]
- Robinson, J.M.; Pasternak, Z.; Mason, C.E.; Elhaik, E. Forensic applications of microbiomics: A review. Front. Microbiol. 2021, 11, 608101. [Google Scholar] [CrossRef] [PubMed]
- Mason-Buck, G.; Graf, A.; Elhaik, E.; Robinson, J.; Pospiech, E.; Oliveira, M.; Moser, J.; Lee, P.K.H.; Githae, D.; Ballard, D.; et al. DNA Based Methods in Intelligence—Moving Towards Metagenomics. Preprints 2020, 2020020158. [Google Scholar]
- Schmedes, S.E.; Sajantila, A.; Budowle, B. Expansion of microbial forensics. J. Clin. Microbiol. 2016, 54, 1964–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, A.C.; Alvarez, M.L.; Davis, C.P.; Guzmán, E.; Han, Y.; Way, L.; Walichiewicz, P.; Silva, D.; Pham, N.; Caves, G.; et al. Developmental validation of the MiSeq FGx forensic genomics system for targeted next-generation sequencing in forensic DNA casework and database laboratories. Forensic Sci. Int. Genet. 2017, 28, 52–70. [Google Scholar] [CrossRef] [Green Version]
- Günther, B.; Jourdain, E.; Rubincam, L.; Karoliussen, R.; Cox, S.L.; Arnaud Haond, S. Feces DNA analyses track the rehabilitation of a free-ranging beluga whale. Sci. Rep. 2022, 12, 6412. [Google Scholar] [CrossRef]
- Székely, D.; Corfixen, N.L.; Mørch, L.L.; Knudsen, S.W.; McCarthy, M.L.; Teilmann, J.; Heide-Jørgensen, M.P.; Olsen, M.T. Environmental DNA captures the genetic diversity of bowhead whales (Balaena mysticetus) in West Greenland. Environ. DNA 2021, 3, 248–260. [Google Scholar] [CrossRef]
- Cordier, T.; Forster, D.; Dufresne, Y.; Martins, C.I.; Stoeck, T.; Pawlowski, J. Supervised machine learning outperforms taxonomy-based environmental DNA metabarcoding applied to biomonitoring. Mol. Ecol. Resour. 2018, 18, 1381–1391. [Google Scholar] [CrossRef]
- Habtom, H.; Pasternak, Z.; Matan, O.; Azulay, C.; Gafny, R.; Jurkevitch, E. Applying microbial biogeography in soil forensics. Forensic Sci. Int. Genet. 2019, 38, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Jesmok, E.M.; Hopkins, J.M.; Foran, D.R. Next-generation sequencing of the bacterial 16S rRNA gene for forensic soil comparison: A feasibility study. J. Forensic Sci. 2016, 61, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Chase, J.; Fouquier, J.; Zare, M.; Sonderegger, D.L.; Knight, R.; Kelley, S.T.; Siegel, J.; Caporaso, J.G. Geography and location are the primary drivers of office microbiome composition. MSystems 2016, 1, e00022-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanachai, A.; Katekeaw, S.; Lomthaisong, K. Forensic soil investigation from the 16S rDNA profiles of soil bacteria obtained by denaturing gradient gel electrophoresis. Chiang Mai J. Sci. 2016, 43, 748–755. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Zorraquino, V.; Tagkopoulos, I. Microbial forensics: Predicting phenotypic characteristics and environmental conditions from large-scale gene expression profiles. PLoS Comput. Biol. 2015, 11, e1004127. [Google Scholar] [CrossRef]
- Cordier, T.; Lanzén, A.; Apothéloz-Perret-Gentil, L.; Stoeck, T.; Pawlowski, J. Embracing environmental genomics and machine learning for routine biomonitoring. Trends Microbiol. 2019, 27, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, K.; Davis, J.; Strom, M.; Werner, C. NOAA’Omics Strategy: Strategic Application of Transformational Tools; National Oceanic and Atmospheric Administration: Washington, DC, USA, 2020. [Google Scholar] [CrossRef]
- Quinn, T.P.; Erb, I.; Richardson, M.F.; Crowley, T.M. Understanding sequencing data as compositions: An outlook and review. Bioinformatics 2018, 34, 2870–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.L.; Zascavage, R.R.; Sedlazeck, F.J.; Planz, J.V. Potential applications of nanopore sequencing for forensic analysis. Forensic Sci. Rev. 2020, 32, 23–54. [Google Scholar] [PubMed]
- The MetaSUB International Consortium. The metagenomics and metadesign of the subways and urban biomes (MetaSUB) international consortium inaugural meeting report. Microbiome 2016, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, C. Decoding the Cryptic Metagenome: A Deep Dive into Gene Clusters and Taxonomy of Microbiome. Ph.D. Dissertation, Weill Medical College of Cornell University, New York, NY, USA, 2020. Order No. 27837631. Available online: https://www.proquest.com/dissertations-theses/decoding-cryptic-metagenome-deep-dive-into-gene/docview/2404392059/se-2 (accessed on 27 August 2022).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Microbiome, metagenomics, and high-dimensional compositional data analysis. Annu. Rev. Stat. Its Appl. 2015, 2, 73–94. [Google Scholar] [CrossRef]
- Network, B. Toward a National Biomonitoring System. J. Environ. Health 2013, 75, 119–212. [Google Scholar]
- Goallec, A.L.; Tierney, B.T.; Luber, J.M.; Cofer, E.M.; Kostic, A.D.; Patel, C.J. A systematic machine learning and data type comparison yields metagenomic predictors of infant age, sex, breastfeeding, antibiotic usage, country of origin, and delivery type. PLoS Comput. Biol. 2020, 16, e1007895. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine learning meta-analysis of large metagenomic datasets: Tools and biological insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [Green Version]
- Denisko, D.; Hoffman, M.M. Classification and interaction in random forests. Proc. Natl. Acad. Sci. USA 2018, 115, 1690–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couronné, R.; Probst, P.; Boulesteix, A.L. Random forest versus logistic regression: A large-scale benchmark experiment. BMC Bioinform. 2018, 19, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statnikov, A.; Henaff, M.; Narendra, V.; Konganti, K.; Li, Z.; Yang, L.; Pei, Z.; Blaser, M.J.; Aliferis, C.F.; Alekseyenko, A.V. A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, A.B.R.; Ounit, R.; Afshinnekoo, E.; Prill, R.J.; Hénaff, E.; Alexander, N.; Minot, S.S.; Danko, D.; Foox, J.; Ahsanuddin, S.; et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017, 18, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bietz, M.J.; Lee, C.P. Collaboration in metagenomics: Sequence databases and the organization of scientific work. In ECSCW 2009; Springer: London, UK, 2009; pp. 243–262. [Google Scholar]
- Tierney, B.T.; Tan, Y.; Kostic, A.D.; Patel, C.J. Gene-level metagenomic architectures across diseases yield high-resolution microbiome diagnostic indicators. Nat. Commun. 2021, 12, 2907. [Google Scholar] [CrossRef]
- Tierney, B.T.; Tan, Y.; Yang, Z.; Shui, B.; Walker, M.J.; Kent, B.M.; Kostic, A.D.; Patel, C.J. Systematically assessing microbiome–disease associations identifies drivers of inconsistency in metagenomic research. PLoS Biol. 2022, 20, e3001556. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Xu, Z.Z.; Bouslimani, A.; Dorrestein, P.; Carter, D.O.; Knight, R. Microbiome tools for forensic science. Trends Biotechnol. 2017, 35, 814–823. [Google Scholar] [CrossRef]
- McCarty, L.S.; Power, M.; Munkittrick, K.R. Bioindicators versus biomarkers in ecological risk assessment. Hum. Ecol. Risk Assess. 2002, 8, 159–164. [Google Scholar] [CrossRef]
- Butler, D.; Mozsary, C.; Meydan, C.; Foox, J.; Rosiene, J.; Shaiber, A.; Danko, D.; Afshinnekoo, E.; MacKay, M.; Sedlazeck, F.J.; et al. Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 2021, 12, 1660. [Google Scholar] [CrossRef]
- Chng, K.R.; Li, C.; Bertrand, D.; Ng, A.H.Q.; Kwah, J.S.; Low, H.M.; Tong, C.; Natrajan, M.; Zhang, M.H.; Xu, L.; et al. Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat. Med. 2020, 26, 941–951. [Google Scholar] [CrossRef]
- Afshinnekoo, E.; Bhattacharya, C.; Burguete-García, A.; Castro-Nallar, E.; Deng, Y.; Desnues, C.; Dias-Neto, E.; Elhaik, E.; Iraola, G.; Jang, S.; et al. COVID-19 drug practices risk antimicrobial resistance evolution. Lancet Microbe 2021, 2, e135–e136. [Google Scholar] [CrossRef]
- Piro, V.C.; Matschkowski, M.; Renard, B.Y. MetaMeta: Integrating metagenome analysis tools to improve taxonomic profiling. Microbiome 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yesson, C.; Brewer, P.W.; Sutton, T.; Caithness, N.; Pahwa, J.S.; Burgess, M.; Gray, W.A.; White, R.J.; Jones, A.C.; Bisby, F.A.; et al. How global is the global biodiversity information facility? PLoS ONE 2007, 2, e1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrêa, F.B.; Saraiva, J.P.; Stadler, P.F.; da Rocha, U.N. TerrestrialMetagenomeDB: A public repository of curated and standardized metadata for terrestrial metagenomes. Nucleic Acids Res. 2020, 48, D626–D632. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.A.; Bhattacharya, C.; Ryon, K.; Meierovich, S.; Shaaban, H.; Westfall, D.; Mohammad, R.; Kuchin, K.; Afshinnekoo, E.; Danko, D.C.; et al. The microbe directory v2. 0: An expanded database of ecological and phenotypical features of microbes. BioRxiv 2019. [Google Scholar] [CrossRef]
- Danko, D.C.; Sierra, M.A.; Benardini, J.N.; Guan, L.; Wood, J.M.; Singh, N.; Seuylemezian, A.; Butler, D.J.; Ryon, K.; Kuchin, K.; et al. A comprehensive metagenomics framework to characterize organisms relevant for planetary protection. Microbiome 2021, 9, 82. [Google Scholar] [CrossRef]
- Arenas, M.; Pereira, F.; Oliveira, M.; Pinto, N.; Lopes, A.M.; Gomes, V.; Carracedo, A.; Amorim, A. Forensic genetics and genomics: Much more than just a human affair. PLoS Genet. 2017, 13, e1006960. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzosa, E.A.; Huang, K.; Meadow, J.F.; Gevers, D.; Lemon, K.P.; Bohannan, B.J.; Huttenhower, C. Identifying personal microbiomes using metagenomic codes. Proc. Natl. Acad. Sci. USA 2015, 112, E2930–E2938. [Google Scholar] [CrossRef] [PubMed]
- Javan, G.T.; Finley, S.J.; Abidin, Z.; Mulle, J.G. The thanatomicrobiome: A missing piece of the microbial puzzle of death. Front. Microbiol. 2016, 7, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.A.; Sanidad, K.Z.; Lucotti, S.; Lieber, C.M.; Cox, R.M.; Ananthanarayanan, A.; Basu, S.; Chen, J.; Shan, M.; Amirm, M.; et al. Gut microbiota-derived metabolites confer protection against SARS-CoV-2 infection. Gut Microbes 2022, 14, 2105609. [Google Scholar] [CrossRef]
- Basu, S.; Liu, C.; Zhou, X.K.; Nishiguchi, R.; Ha, T.; Chen, J.; Johncilla, M.; Yantiss, R.K.; Montrose, D.C.; Dannenberg, A.J. GLUT5 is a determinant of dietary fructose-mediated exacerbation of experimental colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2021, 321, G232–G242. [Google Scholar] [CrossRef]
- Nishiguchi, R.; Basu, S.; Staab, H.A.; Ito, N.; Zhou, X.K.; Wang, H.; Ha, T.; Johncilla, M.; Yantiss, R.K.; Montrose, D.C.; et al. Dietary interventions to prevent high-fructose diet–associated worsening of colitis and colitis-associated tumorigenesis in mice. Carcinogenesis 2021, 42, 842–852. [Google Scholar] [CrossRef]
- Meydan, C.; Afshinnekoo, E.; Rickard, N.; Daniels, G.; Kunces, L.; Hardy, T.; Lili, L.; Pesce, S.; Jacobson, P.; Mason, C.E.; et al. Improved gastrointestinal health for irritable bowel syndrome with metagenome-guided interventions. Precis. Clin. Med. 2020, 3, 136–146. [Google Scholar] [CrossRef]
- Schmidt, C. Living in a microbial world. Nat. Biotechnol. 2017, 35, 401–404. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Machine Learning Methods | Defined Parameters |
|---|---|
| AdaBoost | n_estimators = 1000 (User Defined), learning_rate = 4 |
| Decision Tree | - |
| Extra Tree | n_estimators = 1000 (User Defined), criterion = ‘entropy’ |
| Gaussian Naive Bayes | - |
| K-nearest neighbors | n_neighbors = 21 (User Defined) |
| Linear Discriminant Analysis | - |
| Linear Support Vector Classifier | kernel = ‘linear’, probability = True |
| Logistic Regression | solver = ‘lbfgs’, C = 1e5, max_iter = 1,000,000 |
| Voting Classifier | voting = ”soft” |
| Random Forest Classifier | n_estimators = 1000 (User Defined), criterion = “entropy”, bootstrap = True, warm_start = True |
| Support Vector Machine Classifier | gamma = ‘scale’, decision_function_shape = ‘ovo’, kernel = “rbf”, probability = True |
| Preprocessing Method | |||||||
|---|---|---|---|---|---|---|---|
| Classifier | Binary | CLR | Multiplicative Inverse | Raw | Standard Scalar | Total-Sum | Standard Deviation |
| Extra tree | 0.812 | 0.775 | 0.776 | 0.809 | 0.812 | 0.771 | 0.018328 |
| Linear SVC | 0.889 | 0.893 | 0.581 | 0.89 | 0.883 | 0.562 | 0.1499 |
| Logistic Regression | 0.8952 | 0.89 | 0.879 | 0.895 | 0.8856 | 0.881 | 0.00617 |
| Random Forest | 0.781 | 0.734 | 0.778 | 0.778 | 0.779 | 0.735 | 0.02126 |
| SVM | 0.834 | 0.845 | 0.79 | 0.833 | 0.792 | 0.798 | 0.02226 |
| Feature | #Number of Variables | Desc. of Feature | 10-Fold Av. Accuracy (%) | Leave One Group out (City) Av. Accuracy (%) | Leave One Group Out (Continent) Av. Accuracy (%) | Random Chance (%) |
|---|---|---|---|---|---|---|
| City climate | 13 | Categorized climate Types | 93.0 ± 0.0168 | 34.5 ± 0.3537 | 12.7 ± 0.1338 | 7.69 |
| Coastal | 3 | Coastal city and altitude for non-coastal city | 90.9 ± 0.0123 | 59.1 ± 0.3197 | 40.8 ± 0.1323 | 33.33 |
| Coastal City | 2 | Binary values | 90.9 ± 0.0108 | 52.4 ± 0.2409 | 40.5 ± 0.1009 | 50 |
| Location Type | 68 | Location for collection | 85.7 ± 0.0216 | 44.2 ± 0.3358 | 40.6 ± 0.3221 | 1.47 |
| Surface | 460 | Type of surface | 47.2 ± 0.0177 | 8.7 ± 0.1107 | 3.8 ± 0.0253 | 0.22 |
| Surface Material | 115 | Standardizeding the “Surface” parameter | 54.3 ± 0.0178 | 24.4 ± 0.1920 | 32.5 ± 0.2603 | 0.87 |
| Surface Ontology (Fine) | 6 | Type of surface: stone, biological, etc | 66.2 ± 0.0248 | 50.3 ± 0.2495 | 61.8 ± 0.1532 | 16.67 |
| Surface Ontology (Coarse) | 3 | Surface permeability/ Control | 85.4 ± 0.0127 | 87.1 ± 0.1722 | 83.0 ± 0.0980 | 33.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, C.; Tierney, B.T.; Ryon, K.A.; Bhattacharyya, M.; Hastings, J.J.A.; Basu, S.; Bhattacharya, B.; Bagchi, D.; Mukherjee, S.; Wang, L.; et al. Supervised Machine Learning Enables Geospatial Microbial Provenance. Genes 2022, 13, 1914. https://doi.org/10.3390/genes13101914
Bhattacharya C, Tierney BT, Ryon KA, Bhattacharyya M, Hastings JJA, Basu S, Bhattacharya B, Bagchi D, Mukherjee S, Wang L, et al. Supervised Machine Learning Enables Geospatial Microbial Provenance. Genes. 2022; 13(10):1914. https://doi.org/10.3390/genes13101914
Chicago/Turabian StyleBhattacharya, Chandrima, Braden T. Tierney, Krista A. Ryon, Malay Bhattacharyya, Jaden J. A. Hastings, Srijani Basu, Bodhisatwa Bhattacharya, Debneel Bagchi, Somsubhro Mukherjee, Lu Wang, and et al. 2022. "Supervised Machine Learning Enables Geospatial Microbial Provenance" Genes 13, no. 10: 1914. https://doi.org/10.3390/genes13101914