

Classification and Prognosis Analysis of Pancreatic Cancer Based on DNA Methylation Profile and Clinical Information
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition of DNA Methylation Data and Clinical Information of Patients with PAAD
2.2. Screening and Analysis of Differentially Methylated Genes
2.3. Identification of Methylation Markers Associated with Prognosis
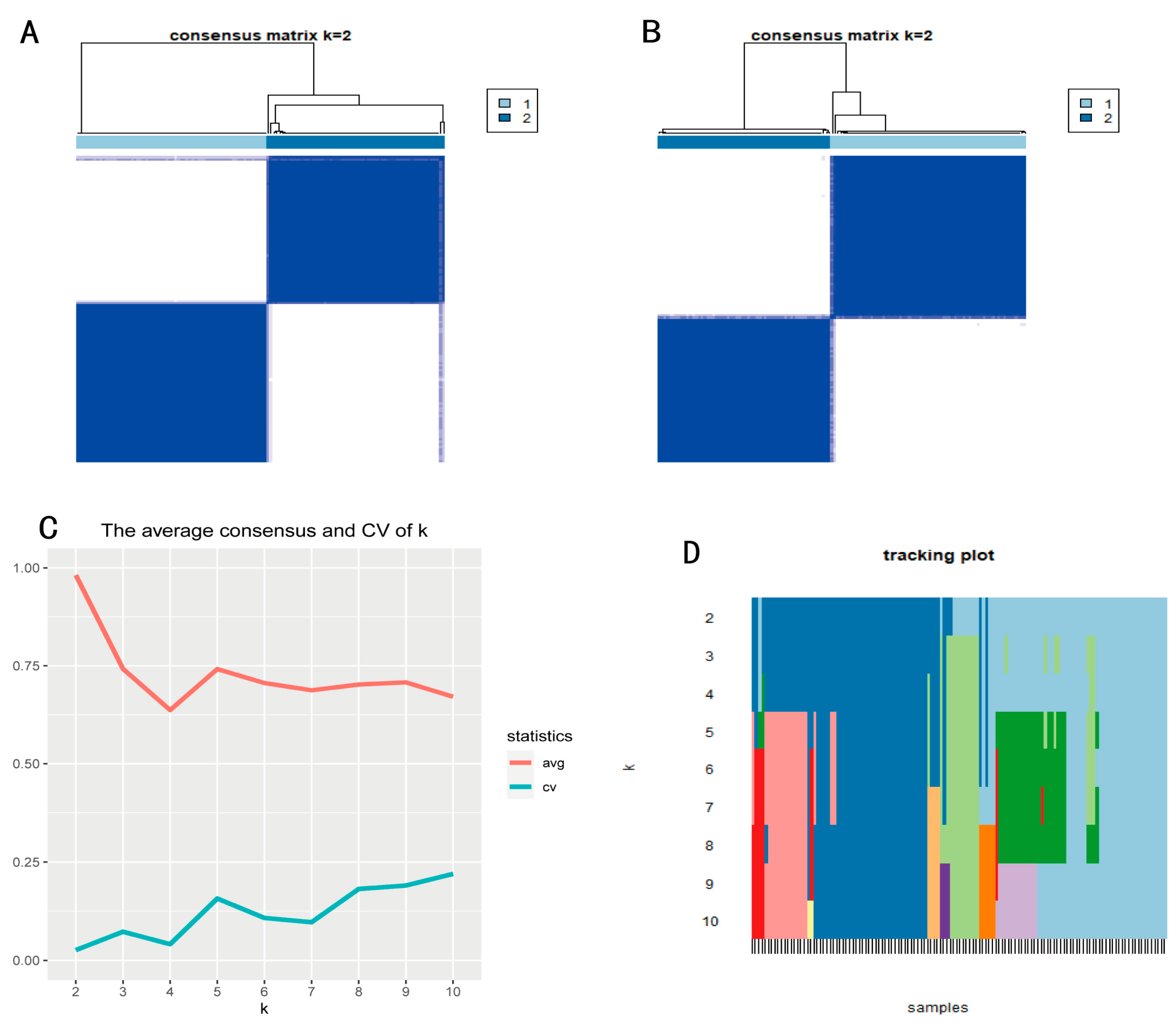
2.4. Subtype Recognition
2.5. Identification of Subtype Specific Marker Genes
2.6. Construction of a Prognosis Prediction Model for PAAD Typing
2.7. Expression Differences According to DNA Methylation Subtype
2.8. Analysis of Immune Cell Abundance in DNA Methylation Subtypes
3. Results
3.1. Identification of Methylation Markers Associated with Prognosis in PAAD
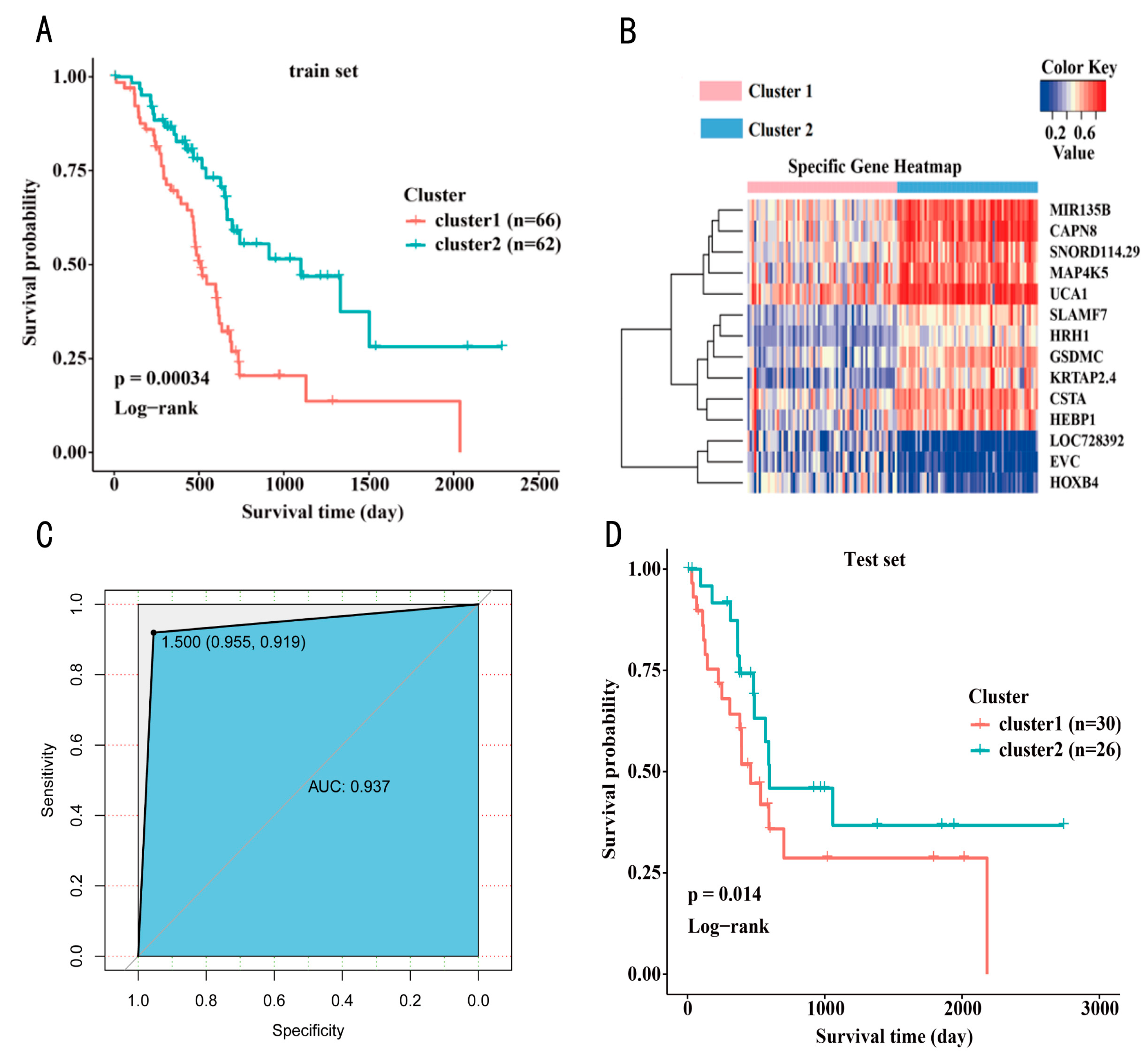
3.2. Construction of a Prognosis Prediction Model for PAAD Classification
3.3. Subtype Analysis of the Prognosis Prediction Model Based on PAAD Classification
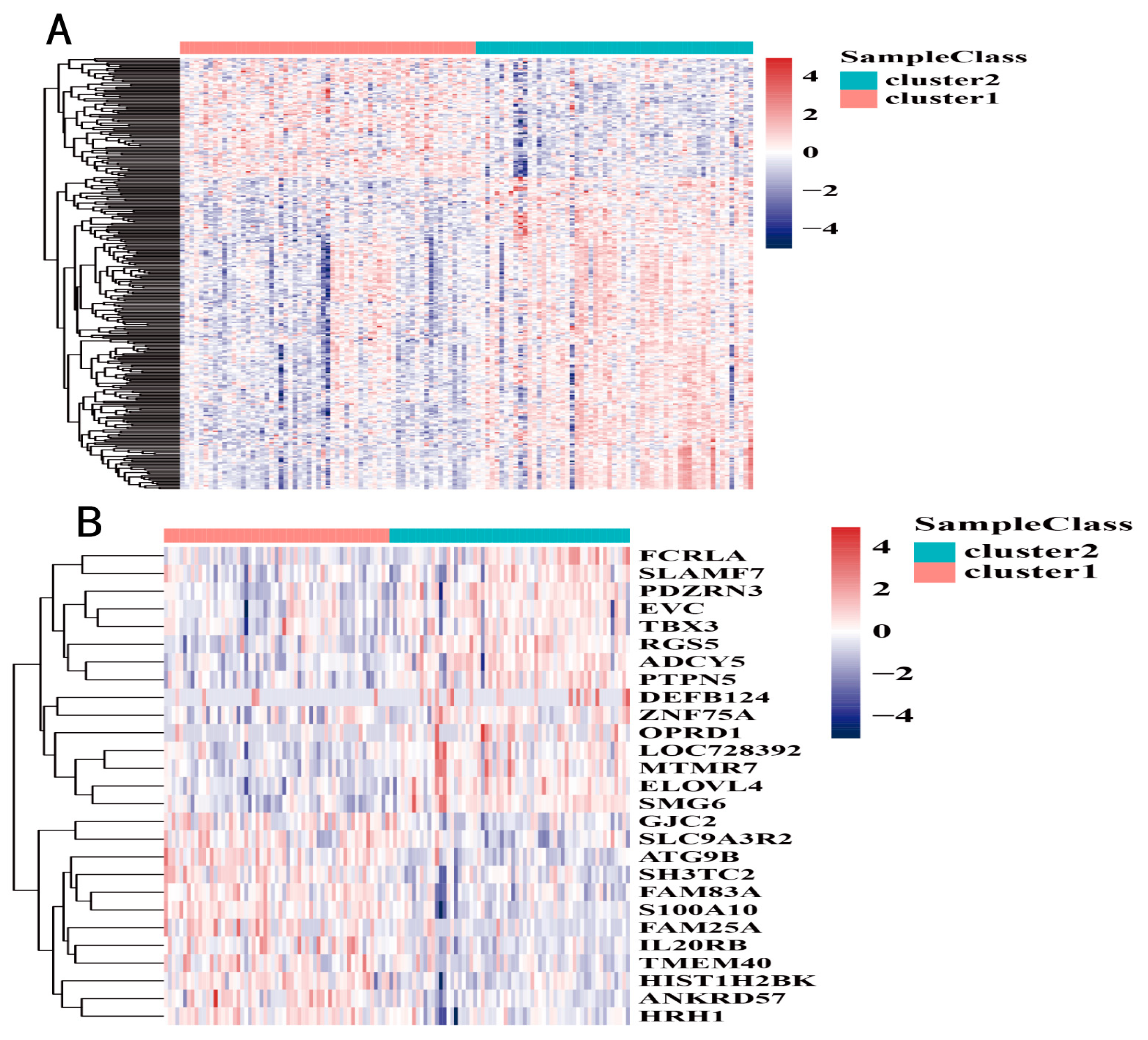
3.4. DNA Methylation Subtype Analysis Based on Transcriptome Data
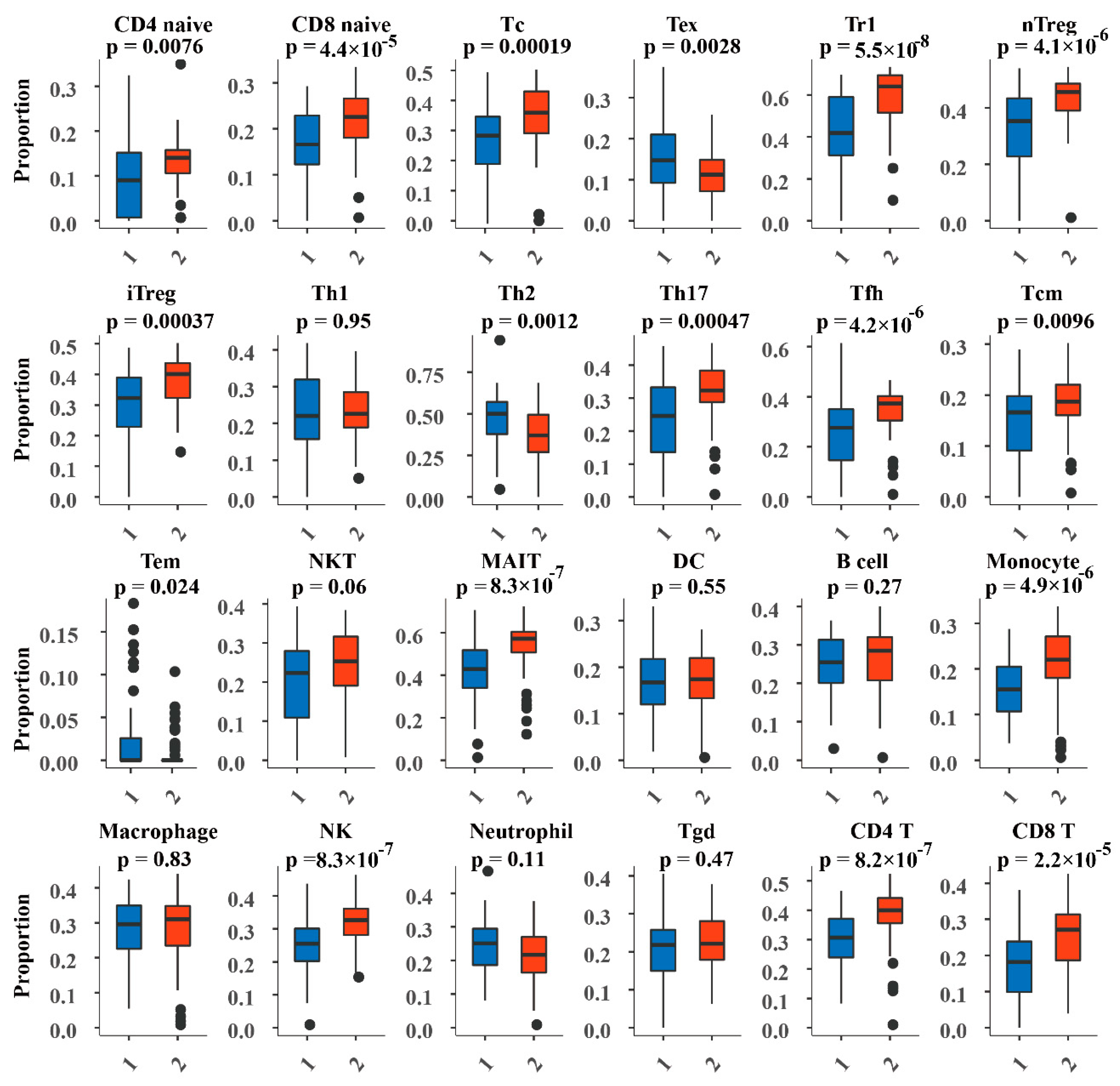
3.5. Abundance of Immune Cells in the DNA Methylated Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Gan, K.A.; Pro, S.C.; Sewell, J.A.; Bass, J.I.F. Identification of Single Nucleotide Non-coding Driver Mutations in Cancer. Front. Genet. 2018, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Deng, T.; Yuan, Y.; Zhang, C.; Zhang, C.; Yao, W.; Wang, C.; Liu, R.; Ba, Y. Identification of Circulating MiR-25 as a Potential Biomarker for Pancreatic Cancer Diagnosis. Cell Physiol. Biochem. 2016, 39, 1716–1722. [Google Scholar] [CrossRef]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epigenetics in breast and prostate cancer. Methods Mol. Biol. 2015, 1238, 425–466. [Google Scholar]
- Mukerjee, S.; Gonzalez-Reymundez, A.; Lunt, S.Y.; Vazquez, A.I. DNA Methylation and Gene Expression with Clinical Covariates Explain Variation in Aggressiveness and Survival of Pancreatic Cancer Patients. Cancer Investig. 2020, 38, 502–506. [Google Scholar] [CrossRef]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Fleischer, T.; Frigessi, A.; Johnson, K.C.; Edvardsen, H.; Touleimat, N.; Klajic, J.; Riis, M.L.; Haakensen, V.D.; Wärnberg, F.; Naume, B.; et al. Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014, 15, 435. [Google Scholar]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Gao, J.; Kwan, P.W.; Shi, D. Sparse kernel learning with LASSO and Bayesian inference algorithm. Neural. Netw. 2010, 23, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Z.; Tian, L.; Zhou, C.; He, M.Y.; Gao, Y.-B.; Wang, S.; Zhou, F.; Shi, S.; Feng, X.; et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut 2014, 63, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Tuti, T.; Agweyu, A.; Mwaniki, P.; Peek, N.; English, M.; Clinical Information Network Author Group. An exploration of mortality risk factors in non-severe pneumonia in children using clinical data from Kenya. BMC Med. 2017, 15, 201. [Google Scholar]
- Yu, K.H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Ré, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 12474. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. BioinforImatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, H.; Lv, J.; Xiao, X.; Zhu, J.; Liu, X.; Su, J.; Li, X.; Wu, Q.; Wang, F.; et al. QDMR: A quantitative method for identification of differentially methylated regions by entropy. Nucleic Acids Res. 2011, 39, e58. [Google Scholar] [CrossRef] [Green Version]
- Brodin, P.; Davis, M.M. Human immune system variation. Nat. Rev. Immunol. 2017, 17, 21–29. [Google Scholar] [CrossRef]
- Miao, Y.R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.Y.; Wang, H.; Guo, A.Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and its Application in Cancer Immunotherapy. Adv. Sci. 2020, 7, 1902880. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Nogueira, P.; Noguera-Castells, A.; Fuster, G.; Recalde-Percaz, L.; Moragas, N.; López-Plana, A.; Enreig, E.; Jauregui, P.; Carbó, N.; Almendro, V.; et al. Histamine receptor 1 inhibition enhances antitumor therapeutic responses through extracellular signal-regulated kinase (ERK) activation in breast cancer. Cancer Lett. 2018, 424, 70–83. [Google Scholar] [CrossRef]
- O’Connell, P.; Hyslop, S.; Blake, M.K.; Godbehere, S.; Amalfitano, A.; Aldhamen, Y.A. SLAMF7 Signaling Reprograms T Cells toward Exhaustion in the Tumor Microenvironment. J. Immunol. 2021, 206, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Gu, Y.; Zhu, J.; Ci, C.; Guo, Z.; Chen, C.; Wei, Y.; Lv, W.; Liu, H.; et al. Specific breast cancer prognosis-subtype distinctions based on DNA methylation patterns. Mol. Oncol. 2018, 12, 1047–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Y.; Wang, Y.; Zhang, Y. Identification of prognostic signature of non-small cell lung cancer based on TCGA methylation data. Sci. Rep. 2020, 10, 8575. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.K.; Guda, C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 2017, 8, 28990–29012. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, W.; Spieker, N.; van Roy, N.; De Boulle, K.; De Paepe, A.; Willems, P.; van Hul, W.; Versteeg, R.; Speleman, F. Refined physical mapping and genomic structure of the EXTL1 gene. Cytogenet. Cell Genet. 1999, 86, 267–270. [Google Scholar] [CrossRef]
- Redon, S.; Reichenbach, P.; Lingner, J. Protein RNA and protein protein interactions mediate association of human EST1A/SMG6 with telomerase. Nucleic Acids Res. 2007, 35, 7011–7022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inozume, T.; Mitsui, H.; Okamoto, T.; Matsuzaki, Y.; Kawakami, Y.; Shibagaki, N.; Shimada, S. Dendritic cells transduced with autoantigen FCRLA induce cytotoxic lymphocytes and vaccinate against murine B-cell lymphoma. J. Investig. Dermatol. 2007, 127, 2818–2822. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Entropy | Cluster1 | Cluster2 |
|---|---|---|---|
| CAPN8 | 8.4890 | 0.4903 | 0.7586 |
| MIR135B | 8.6130 | 0.5052 | 0.7505 |
| LOC728392 | 8.7880 | 0.3407 | 0.1245 |
| KRTAP2.4 | 8.8170 | 0.3170 | 0.5288 |
| GSDMC | 8.8670 | 0.3958 | 0.6001 |
| HEBP1 | 8.8730 | 0.4158 | 0.6190 |
| SNORD114.29 | 8.9020 | 0.4707 | 0.6698 |
| MAP4K5 | 8.9140 | 0.4968 | 0.6941 |
| EVC | 8.9250 | 0.3252 | 0.1295 |
| UCA1 | 8.9460 | 0.5976 | 0.7903 |
| CSTA | 9.0130 | 0.4692 | 0.6528 |
| SLAMF7 | 9.0760 | 0.3656 | 0.5409 |
| HOXB4 | 9.1070 | 0.3848 | 0.2134 |
| HRH1 | 9.1590 | 0.3117 | 0.4766 |
| Prediction Cluster1 | Prediction Cluster2 | |
|---|---|---|
| Cluster1 | 63 | 3 |
| Cluster2 | 5 | 57 |
| Gene | Fold Change | Adjusted p-Value |
|---|---|---|
| HRH1 | 2.3112 | |
| LOC728392 | 0.3919 | |
| SLAMF7 | 0.3007 | |
| EVC | 0.3859 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, X.; Lin, X.; Cai, L.; Wang, Y.; Chang, Z. Classification and Prognosis Analysis of Pancreatic Cancer Based on DNA Methylation Profile and Clinical Information. Genes 2022, 13, 1913. https://doi.org/10.3390/genes13101913
Li X, Zhang X, Lin X, Cai L, Wang Y, Chang Z. Classification and Prognosis Analysis of Pancreatic Cancer Based on DNA Methylation Profile and Clinical Information. Genes. 2022; 13(10):1913. https://doi.org/10.3390/genes13101913
Chicago/Turabian StyleLi, Xin, Xuan Zhang, Xiangyu Lin, Liting Cai, Yan Wang, and Zhiqiang Chang. 2022. "Classification and Prognosis Analysis of Pancreatic Cancer Based on DNA Methylation Profile and Clinical Information" Genes 13, no. 10: 1913. https://doi.org/10.3390/genes13101913