4.1. Prevalence of Lynch Syndrome and Frequency of MMR Pathogenic Variants
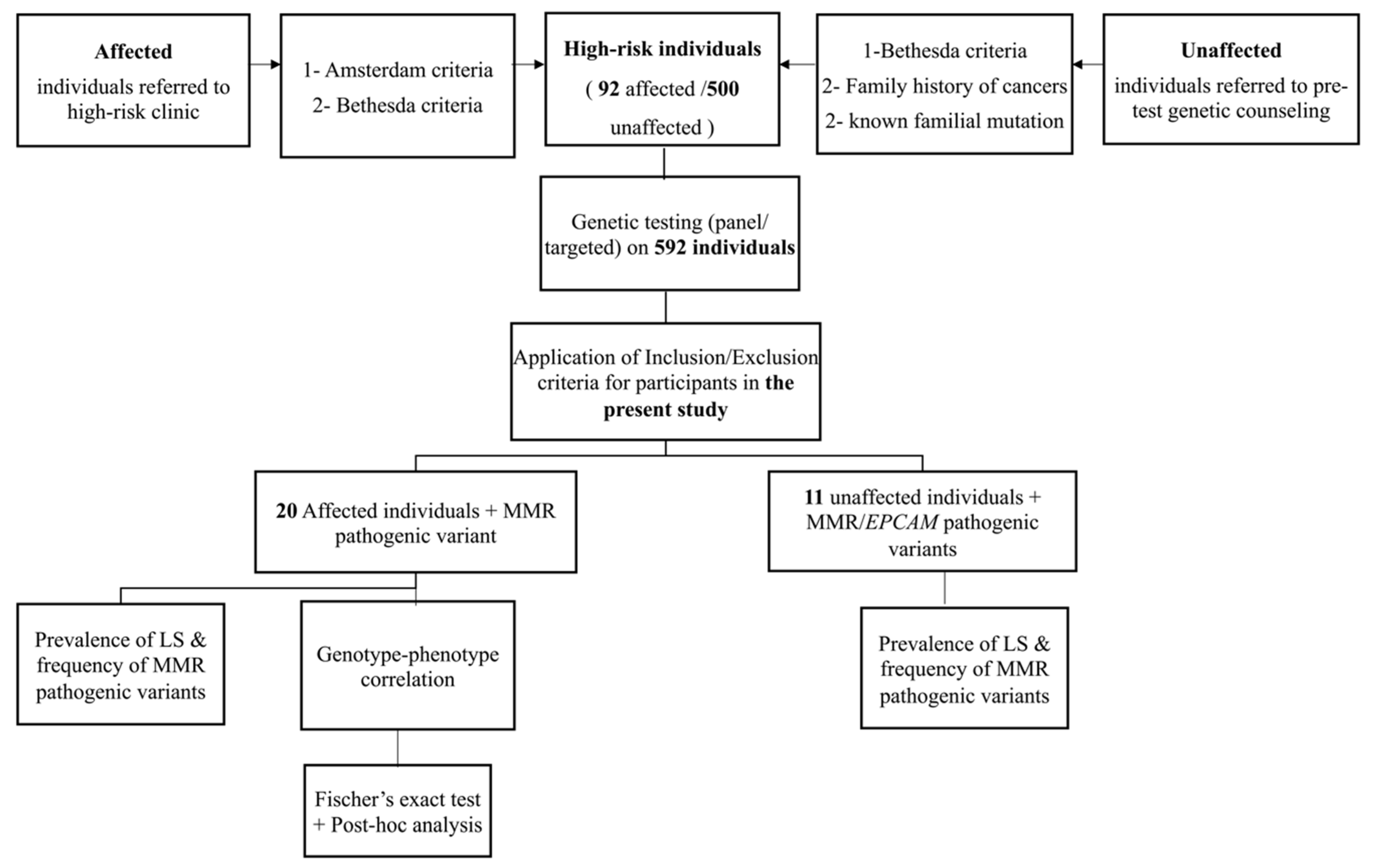
To our knowledge, this is the first study to investigate the prevalence and genotype-phenotype correlation of Lynch syndrome in the state of Qatar. We investigated the prevalence of LS, frequency of pathogenic variants in MMR/EPCAM genes, and genotype-phenotype correlation in 31 individuals; 20 were affected with colon cancer and 11 were unaffected.
The prevalence of Lynch Syndrome among our selected CRC patients was found to be 22% (20/92). Compared to the studies from the MENA (Middle East and North Africa) region, this prevalence is less than what has been found in Pakistani selected patients (34.5%) as they have followed a very stringent criteria of selection (at least three relatives affected with LS associated cancers, at least one of them is a first degree relative of the other two, at least 2 different generations with LS associated cancers, individuals with cancer diagnosed at an age younger than 50 years) [
22]. This prevalence of LS found in our patients is however, higher than most of the other reported CRC prevalence in the remaining MENA countries. For instance, in the Kingdom of Saudi Arabia (KSA) the prevalence of LS in CRC patients was 7% [
23] and in Iran, it was 5.5% [
24]. The prevalence of LS in our CRC patients was also higher compared to the prevalence reported by a study in the United States of America (USA) conducted mainly in the white population of Ohio (72/450) 16% [
25].
The prevalence of Lynch syndrome in unaffected high-risk individuals was 2.2% (11/500), this prevalence was lower than that of unaffected high-risk relatives of patients with Lynch syndrome from Columbus, Ohio (USA) (77%; 102/132). Such high prevalence in the Ohio study is due to the fact that it was calculated among mainly first degree relatives of patients with confirmed pathogenic variants in one of the MMR genes and thus the possibility of detecting a pathogenic variant in a first degree relative is 50%. However, this is not the case for the high-risk unaffected individuals in the current study as not all of them had an affected relative confirmed to have pathogenic variants in MMR due to the fact that most didn’t pursue testing which could explain the lower prevalence of Lynch syndrome detected in our study compared to the previous Ohio study. Our approach to evaluate the prevalence of LS in selected high-risk patients/individuals affected or unaffected gives more insights regarding the genetic testing approach mainly for unaffected individuals. In this regard, our findings demonstrate the benefit of panel genetic testing for healthy high-risk individuals based on their family history especially when the types of cancers overlap in multiple genetic syndromes even though no pathogenic variant has been confirmed in relatives. Following, panel genetic testing approach, about 2.2% of unaffected relatives could be positive for any MMR gene variant and their identification could reduce their cancer risk through early surveillance, prophylactic prevention and early detection of cancers [
26].
Among the affected individuals, the most reported genes were
MLH1 and
MSH2 accounting for 90% of the pathogenic variants, which is in agreement with earlier studies from Saudi Arabia [
27] the United States [
28], Finland [
29], Spain [
30] and the results of the international Mismatch Repair Consortium (IMRC) [
15] This high prevalence of
MLH1 and
MSH2 genes in our cohort could also be attributed to the fact that affected patients were referred based on Amsterdam and Bethesda criteria, as families fulfilling these 2 criteria are more likely to harbor pathogenic variants in
MLH1 and
MSH2 genes [
31] which might have resulted in missing some patients with pathogenic variants MSH6 and PMS2 genes. For unaffected LS individuals however, the most reported gene was
PMS2 accounting for 64% followed by
MSH6 and
EPCAM genes with each accounting for 18% of the pathogenic variants. This high frequency of
PMS2 gene is due to the fact that all carriers were members of the same tribe. This finding suggests the presence of a potential tribal variant in
PMS2 gene which has a great clinical benefit as it can facilitate the selection of most of the at-risk individuals belonging to the same tribe for pre-test counseling and early cancer surveillance and pre-implantation genetic diagnosis. Such interventions would prevent further transmission of the pathogenic variant to the future generations. Additionally, the difference in the distribution of the genes between affected and unaffected individuals was statistically significant (
p < 0.005) after Bonferroni correction, and the fact that
MLH1 and
MSH2 genes were not reported in unaffected individuals could be attributed to the high penetrance of these 2 genes and their association with higher cancer risk compared to the remaining MMR and
EPCAM genes which makes them less likely to be detected in unaffected individuals [
32].
With regards to the type of variants in each gene, for affected patients, point mutations and frameshift variants were commonly reported in
MLH1,
MSH2, and
MSH6 genes which goes in line with what has been reported in the Human Gene Mutation Database regarding the most common variant type in each gene (
http://www.hgmd.cf.ac.uk/ac/all.php, accessed on 14 January 2022). For
PMS2 gene, however, one point mutation and one novel large deletion (deletion encompassing exons 6 to 11) were reported (
Table S1). A large deletion (deletion encompassing exons 6 to 11) was observed to be recurrent in 3 affected members of the same tribe in a homozygous state which is consistent with the diagnosis of Constitutional Mismatch Repair Deficiency Syndrome (CMMRD), a childhood-onset syndrome. Nevertheless, it is interesting to note the delay in onset of disease in these affected individuals (23, 25, and 28 years for patients C0021, C0022, and C0023 respectively) (
Table S1). This late-onset could be explained by the fact that CMMRD,
PMS2/MSH6 homozygous pathogenic variants are associated with a later onset phenotype compared to homozygous pathogenic variants in
MLH1/MSH2 genes which result in more aggressive hematological malignancies during young childhood and are associated with a worse prognosis [
33]. A potential explanation for this late age at onset could also be the partial compensation of absent PMS2 by MLH3, which can form a functional heterodimeric protein with MLH1 that has mismatch repair capacity [
34]. However, the surveillance for CMMRD is similar for all affected individuals regardless of which MMR gene was involved [
35].
For unaffected individuals, the same previous recurrent large deletion (deletion encompassing exons 6 to 11) in
PMS2 gene has also been reported in a heterozygous state in 4 unaffected individuals belonging to the same tribe. Generally heterozygous pathogenic variants in
PMS2 display an attenuated Lynch syndrome phenotype consisting of lower penetrance and a later age at onset [
36]. For
EPCAM gene, all pathogenic variants were also large deletions which is in agreement with the nature of the common variants in this gene reported in other studies [
6]. The heterozygous deletion of the entire
EPCAM gene reported in patient U004 (
Table S2) and also previously reported in an individual with Lynch syndrome associated cancer [
37], was reported of unknown significance on cancer risk because, it is known that deletions of 3′ region of
EPCAM gene are associated with silencing of
MSH2 gene through the transcriptional read-through [
38]. However, in the deletion we report in this study, the entire
EPCAM gene is deleted and as a result transcription and transcriptional read-through might not occur, thus
MSH2 gene might not be affected which results in an unknown risk for Lynch Syndrome. However, it is considered pathogenic with respect to Congenital Tufting Enteropathy which is an autosomal recessive condition associated with biallelic pathogenic variants in the
EPCAM gene [
39]. Thereby, in the Qatari population where the consanguinity rate is high (54%) [
40], the chances of serious autosomal recessive childhood-onset conditions such as CMMRD and Congenital Tufting enteropathy is increased especially with the presence of known Tribal variants. This suggests the necessity of implementing testing for targeted tribal variants in pre-marital screening and offering pre-implantation genetic testing for carrier parents to avoid the risk of autosomal recessive conditions associated with being homozygous for pathogenic variants in MMR/
EPCAM genes [
33,
39].
4.2. Genotype-Phenotype Correlation in Affected LS Patients
There was no statistically significant association established between the type of cancer and mutated gene. One previous study found a lower expression of MLH1 protein in right sided colon cancer and a loss of MSH2 protein expression in the left sided colon cancers which might be due to germline mutations in the corresponding genes, however the association was not statistically significant [
41].
Additionally, although it was expected that
MLH1 and
MSH2 genes would be associated with a younger age at onset as compared to
MSH6 and
PMS2 there was no statistically significant correlation (
Table 5)
In line with what has been published in the literature [
7,
16], we did not find a statistically significant correlation between gender, ethnicity, age, tumor location, type of cancer, grade of cancer, lymph vascular, invasiveness, mucinous component, family history, histopathology, and tumor size with the type of mutated gene which might also be explained by our small sample size (
Table 5).
It was expected that carriers of large deletions and frameshift variants would exhibit a more severe phenotype and an earlier age at onset compared to carriers of point mutations [
42]. However, there was no statistical correlation between the type of variant and any of the clinicopathological parameters tested especially after Bonferroni correction for the association of CRC side with the type of mutated variant (
Table 6). These findings are in agreement with the findings of a previous study on lynch syndrome patients from Spain where their correlation of the type of variant with clinicopathological variables did not yield any statistically significant results [
7].
Immunohistochemistry testing was performed on tumors from all affected individuals with colon cancers as a first step (before the referral to the high-risk clinic) to test for the presence of MMR proteins and select individuals at high risk for LS. In our cohort, most of
MLH1 pathogenic variants, (6/9) (66.7%) had a corresponding loss of MlH1 protein expression along with the loss of its heterodimer PMS2. The loss of MLH1 and PMS2 proteins on IHC is expected as MLH1/PMS2 tend to form heterodimers. Regarding
MSH2, (7 variants resulted in a corresponding loss of MSH2 protein expression and its heterodimer MSH6 protein, the loss of MSH2 and MSH6 proteins on IHC is due to the formation of heterodimers between these 2 proteins. However, the loss of MLH1 protein could be explained by the low sensitivity associated with some antibodies used in the IHC technique [
11]. On the other hand, one pathogenic frameshift variant detected in
MSH6 gene resulted in intact MMR proteins expression. One of the limitations of IHC technique is that not all pathogenic MMR variants result in loss of immunoreactivity, for instance, frameshift and truncating variants can interfere with the protein function without altering the antigenic site of the protein [
43], additionally, the interaction between MMR proteins could affect the sensitivity of immunohistochemistry in the detection of individual proteins [
11].
Therefore, regardless of which protein was lost based on IHC, performing Panel testing is recommended for all individuals suspected to have Lynch syndrome [
44] and not strictly meeting Bethesda or Amsterdam criteria. Clinicians should attentively take the personal and family history of the patient to be able to assess their eligibility for genetic testing. Evidently, the adoption of an effective screening program is challenging and is a topic of ongoing debate in the literature [
27].




{kind=link}