
EpiReSIM: A Resampling Method of Epistatic Model without Marginal Effects Using Under-Determined System of Equations

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetics and Modeling
2.2. Calculating of Epistatic Model
2.3. Resampling to Generate Samples
2.4. Generating the Labels of Samples
3. Results
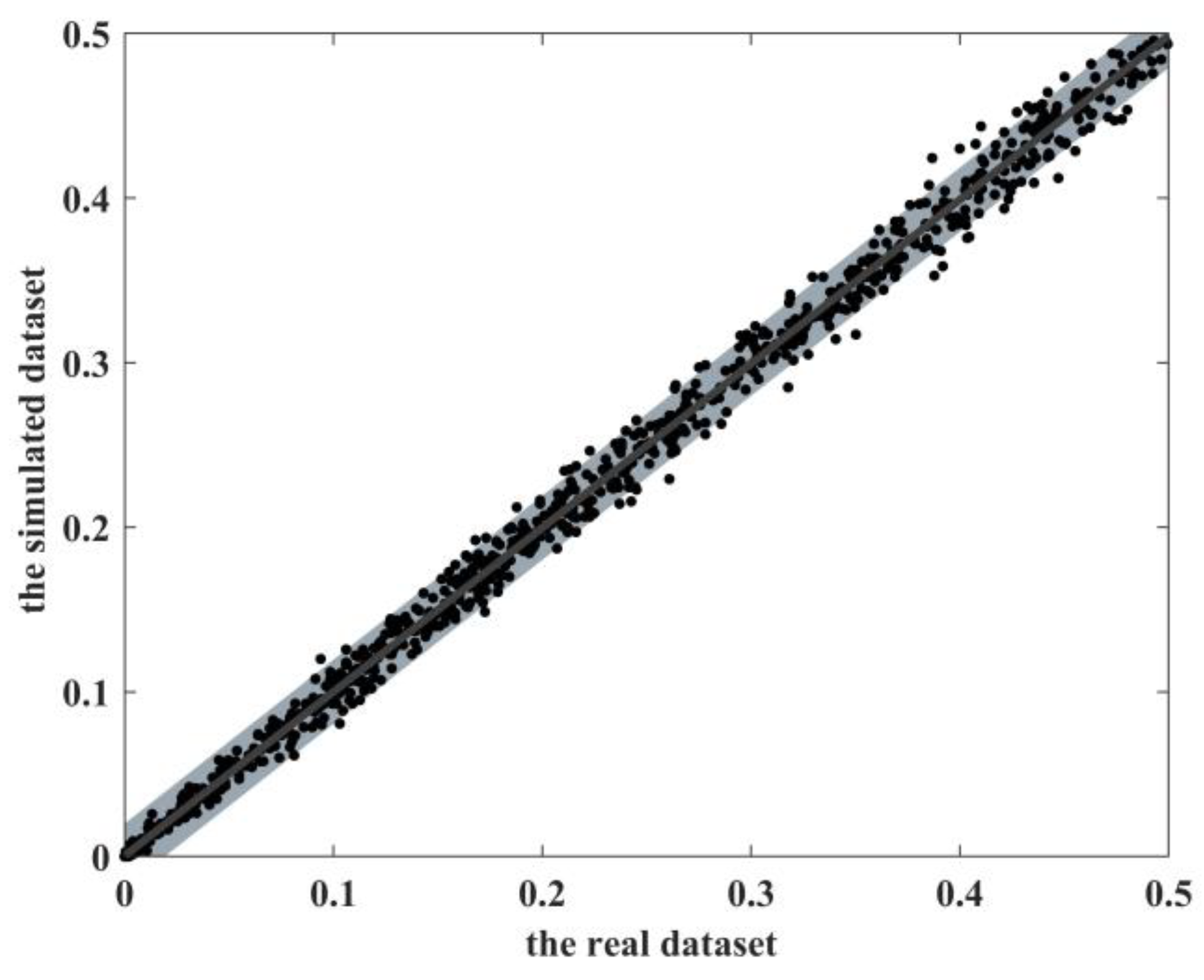
3.1. Preservation of Realistic MAFs
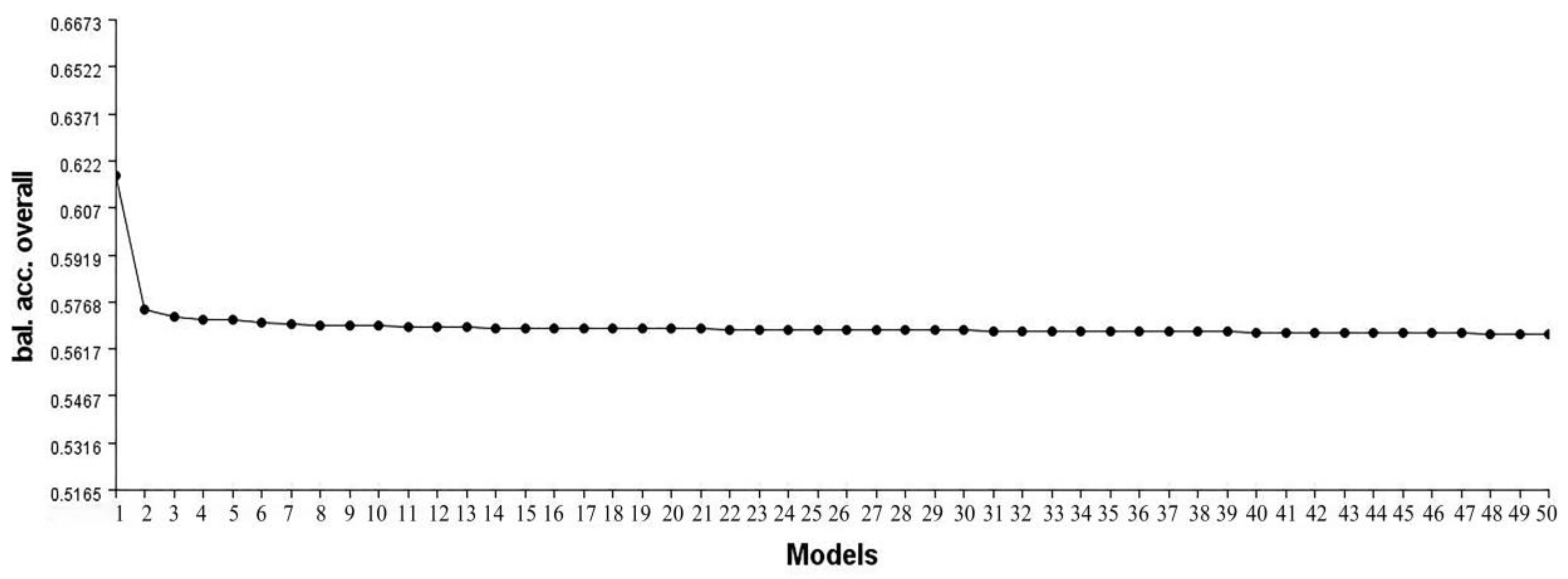
3.2. Accuracy Epistasis Model Embedding
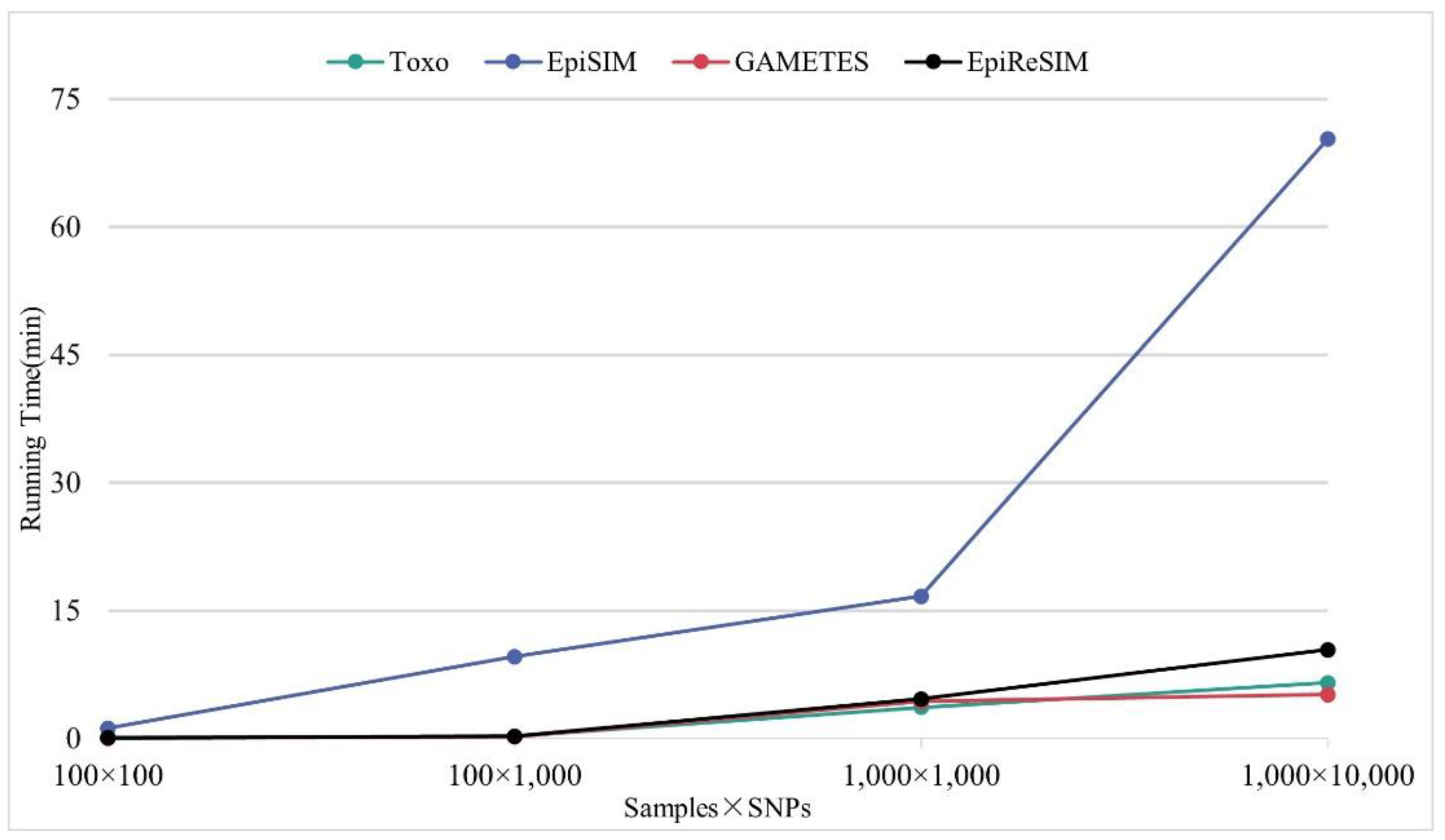
3.3. Acceptable Generating Time
3.4. Comparison with Existing Simulators
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shang, J.; Zhang, J.; Lei, X.; Zhao, W.; Dong, Y. EpiSIM: Simulation of multiple epistasis, linkage disequilibrium patterns and haplotype blocks for genome-wide interaction analysis. Genes Genom. 2013, 35, 305–316. [Google Scholar] [CrossRef]
- Tuo, S. FDHE-IW: A Fast Approach for Detecting High-Order Epistasis in Genome-Wide Case-Control Studies. Genes 2018, 9, 435. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, J.; Liu, J.-X.; Li, S.; Zheng, C.-H. epiACO-a method for identifying epistasis based on ant Colony optimization algorithm. BioData Min. 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, J.; Liu, J.; Li, S. An Improved Ant Colony Optimization Algorithm for the Detection of SNP-SNP Interactions. In Proceedings of the International Conference on Intelligent Computing, Lanzhou, China, 2–5 August 2016; pp. 21–32. [Google Scholar] [CrossRef]
- Escalona, M.; Rocha, S.; Posada, D. A comparison of tools for the simulation of genomic next-generation sequencing data. Nat. Rev. Genet. 2016, 17, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wu, X.; Jiang, R.; Li, Y. Epistatic Module Detection for Case-Control Studies: A Bayesian Model with a Gibbs Sampling Strategy. PLoS Genet. 2009, 5, e1000464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.S. Bayesian inference of epistatic interactions in case-control studies. Nat. Genet. 2007, 39, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Fernández, C.; González-Domínguez, J.; Carvajal-Rodriguez, A.; Martín, M.J. Toxo: A library for calculating penetrance tables of high-order epistasis models. BMC Bioinform. 2020, 21, 138. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, D.B.; Viola, L.; List, M.; Baumbach, J.; Tieri, P.; Kacprowski, T. EpiGEN: An epistasis simulation pipeline. Bioinformatics 2020, 36, 4957–4959. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J. Detecting gene–gene interactions that underlie human diseases. Nat. Rev. Genet. 2009, 10, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Culverhouse, R.; Suarez, B.K.; Lin, J.; Reich, T. A Perspective on Epistasis: Limits of Models Displaying No Main Effect. Am. J. Hum. Genet. 2002, 70, 461–471. [Google Scholar] [CrossRef]
- Urbanowicz, R.J.; Kiralis, J.; Sinnott-Armstrong, N.A.; Heberling, T.; Fisher, J.M.; Moore, J.H. GAMETES: A fast, direct algorithm for generating pure, strict, epistatic models with random architectures. BioData Min. 2012, 5, 16. [Google Scholar] [CrossRef]
- Hartl, D.L.; Clark, A.G.; Clark, A.G. Principles of Population Genetics; Sinauer associates: Sunderland, MA, USA, 1997; Volume 116. [Google Scholar]
- Jing, P.-J.; Shen, H.-B. MACOED: A multi-objective ant colony optimization algorithm for SNP epistasis detection in genome-wide association studies. Bioinformatics 2014, 31, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, F.; Pian, C.; Xu, M.; Kong, L.; Fang, J.; Li, Z.; Zhang, L. EpiMOGA: An Epistasis Detection Method Based on a Multi-Objective Genetic Algorithm. Genes 2021, 12, 191. [Google Scholar] [CrossRef] [PubMed]
- Hough, P.D.; Vavasis, S.A. Complete Orthogonal Decomposition for Weighted Least Squares. SIAM J. Matrix Anal. Appl. 1997, 18, 369–392. [Google Scholar] [CrossRef]
- Broyden, C. The convergence of an algorithm for solving sparse nonlinear systems. Math. Comput. 1971, 25, 285–294. [Google Scholar] [CrossRef]
- Dennis, J.E., Jr.; Schnabel, R.B. Numerical Methods for Unconstrained Optimization and Nonlinear Equations; SIAM: Philadelphia, PA, USA, 1996. [Google Scholar]
- Kelley, C.T. Solving Nonlinear Equations with Newton’s Method; SIAM: Philadelphia, PA, USA, 2003. [Google Scholar]
- Shi, M.; Umbach, D.M.; Wise, A.S.; Weinberg, C.R. Simulating autosomal genotypes with realistic linkage disequilibrium and a spiked-in genetic effect. BMC Bioinform. 2018, 19, 2. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, L.R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-Dimensionality Reduction Reveals High-Order Interactions among Estrogen-Metabolism Genes in Sporadic Breast Cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SNP1 | Marginal Penetrance | ||||
|---|---|---|---|---|---|
| Genotype | |||||
| SNP2 | (0.64) | 0.4865 | 0.9601 | 0.5377 | 0.64 |
| (0.32) | 0.9473 | 0.0042 | 0.8113 | 0.64 | |
| (0.04) | 0.6401 | 0.6065 | 0.9089 | 0.64 | |
| Marginal penetrance | 0.64 | 0.64 | 0.64 | P (D) = 0.64 | |
| SNP1 | Marginal Penetrance | ||||
|---|---|---|---|---|---|
| Genotype | |||||
| SNP2 | P (D|BB) | ||||
| P (D|Bb) | |||||
| P (D|bb) | |||||
| Marginal penetrance | P (D|AA) | P (D|Aa) | P (D|aa) | P (D) | |
| Order | P (D) | SNP Loci | |
|---|---|---|---|
| Model 1 | 2-order | 0.1 | 825, 511 |
| Model 2 | 2-order | 0.2 | 197, 52 |
| Model 3 | 2-order | 0.3 | 687, 74 |
| Model 4 | 2-order | 0.4 | 23, 696 |
| Model 5 | 3-order | 0.3 | 962, 193, 339 |
| Model 6 | 3-order | 0.4 | 461, 755, 428 |
| Model 7 | 4-order | 0.1 | 176, 76, 439, 465 |
| Model 8 | 4-order | 0.2 | 497, 46, 362, 123 |
| Model | Results | Power | ||
|---|---|---|---|---|
| 1000 Samples | 2000 Samples | 4000 Samples | ||
| Model 1 | 511, 825 | 0.15 | 0.80 | 1.00 |
| Model 2 | 52, 197 | 0.20 | 0.90 | 1.00 |
| Model 3 | 74, 687 | 0.70 | 1.00 | 1.00 |
| Model 4 | 23, 696 | 1.00 | 1.00 | 1.00 |
| Model 5 | 193, 339, 962 | 0.45 | 0.95 | 1.00 |
| Model 6 | 428, 461, 755 | 0.25 | 0.80 | 1.00 |
| Model 7 | 76, 176, 439, 465 | 0.75 | 1.00 | 1.00 |
| Model 8 | 46, 123, 362, 497 | 0.80 | 1.00 | 1.00 |
| MAF | P (D) | Time (s) | P (D) | h2 | Time (s) | |
|---|---|---|---|---|---|---|
| 2-order | 0.1, 0.4 | 0.1 | 3.4554 | 0.2 | 0.1 | 3.6100 |
| 0.2, 0.3 | 0.1 | 3.0771 | 0.2 | 0.1 | 3.4776 | |
| 0.1, 0.4 | 0.2 | 2.9695 | 0.1 | 0.05 | 3.2221 | |
| 0.2, 0.3 | 0.2 | 2.8789 | 0.1 | 0.05 | 3.4221 | |
| 3-order | 0.1, 0.4, 0.2 | 0.1 | 4.5305 | 0.2 | 0.05 | 5.9646 |
| 0.2 | 4.0120 | 0.1 | 0.1 | 5.6743 | ||
| 4-order | 0.1, 0.3, 0.2, 0.4 | 0.1 | 8.4997 | 0.1 | 0.05 | 13.3102 |
| 0.15 | 8.9237 | 0.05 | 0.05 | 13.4144 |
| EpiSIM | GAMETES | Toxo | EpiReSIM | |
|---|---|---|---|---|
| preserve MAF of real datasets | × | × | × | √ |
| simulate high-order interactions | × | √ | √ | √ |
| support MAF specification | √ | √ | √ | √ |
| not depend on other software | √ | √ | × | √ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, J.; Cai, X.; Zhang, T.; Sun, Y.; Zhang, Y.; Liu, J.; Guan, B. EpiReSIM: A Resampling Method of Epistatic Model without Marginal Effects Using Under-Determined System of Equations. Genes 2022, 13, 2286. https://doi.org/10.3390/genes13122286
Shang J, Cai X, Zhang T, Sun Y, Zhang Y, Liu J, Guan B. EpiReSIM: A Resampling Method of Epistatic Model without Marginal Effects Using Under-Determined System of Equations. Genes. 2022; 13(12):2286. https://doi.org/10.3390/genes13122286
Chicago/Turabian StyleShang, Junliang, Xinrui Cai, Tongdui Zhang, Yan Sun, Yuanyuan Zhang, Jinxing Liu, and Boxin Guan. 2022. "EpiReSIM: A Resampling Method of Epistatic Model without Marginal Effects Using Under-Determined System of Equations" Genes 13, no. 12: 2286. https://doi.org/10.3390/genes13122286
APA StyleShang, J., Cai, X., Zhang, T., Sun, Y., Zhang, Y., Liu, J., & Guan, B. (2022). EpiReSIM: A Resampling Method of Epistatic Model without Marginal Effects Using Under-Determined System of Equations. Genes, 13(12), 2286. https://doi.org/10.3390/genes13122286