
Unaffected Li-Fraumeni Syndrome Carrier Parent Demonstrates Allele-Specific mRNA Stabilization of Wild-Type TP53 Compared to Affected Offspring


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture, Irradiation, and Harvest of Human Dermal Fibroblasts
2.2. Immunoblot Analysis
2.3. p53 Promoter Binding Activity
2.4. Cytosine Methylation Analysis of TP53 Gene Promoter
2.5. Reverse-Transcribed (RT)-qPCR mRNA Expression
2.6. TP53 mRNA Half-Life
2.7. RT-PCR-RFLP Analysis for TP53 Allele-Specific mRNA Stability
2.8. Statistics
3. Results
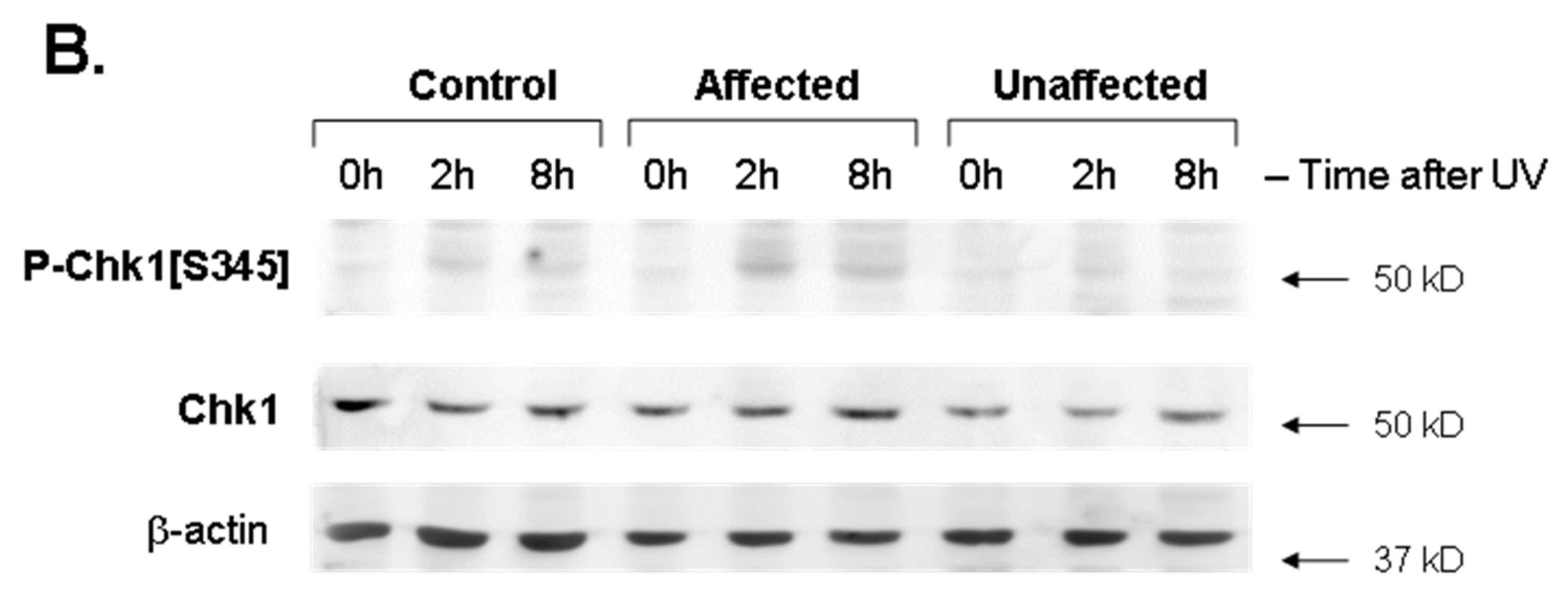
3.1. Phospho-Chk1[S345] Induction Was Significantly Upregulated by UV Exposure in Affected LFS Fibroblasts
3.2. Mutant p53 Protein Expression Was Significantly Reduced in Unaffected LFS Fibroblasts
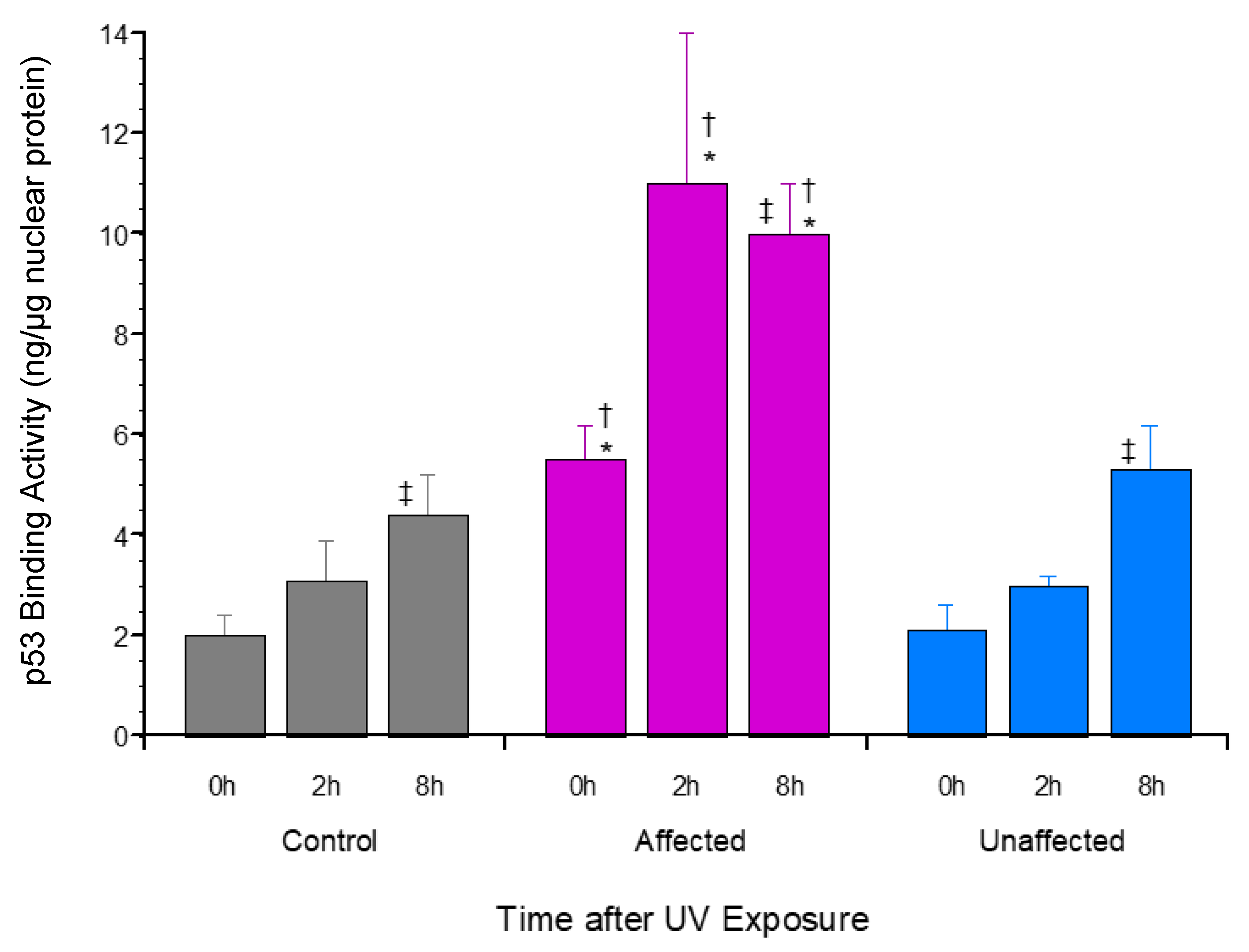
3.3. p53 Promoter Binding Activity Was Significantly Upregulated by UV Exposure in Affected LFS Fibroblasts
3.4. Cytosine Methylation of TP53 Gene Promoters Was Not Detected
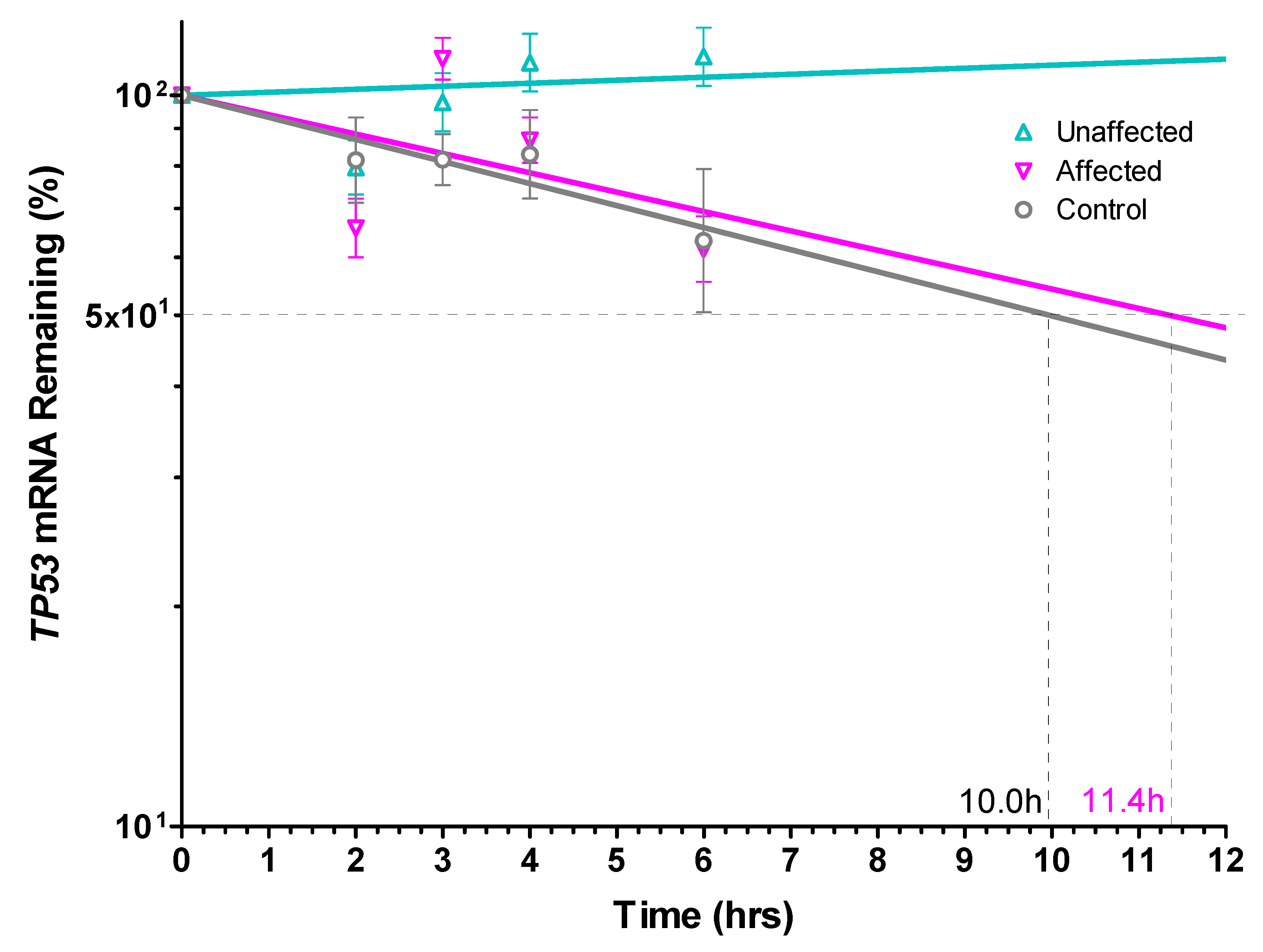
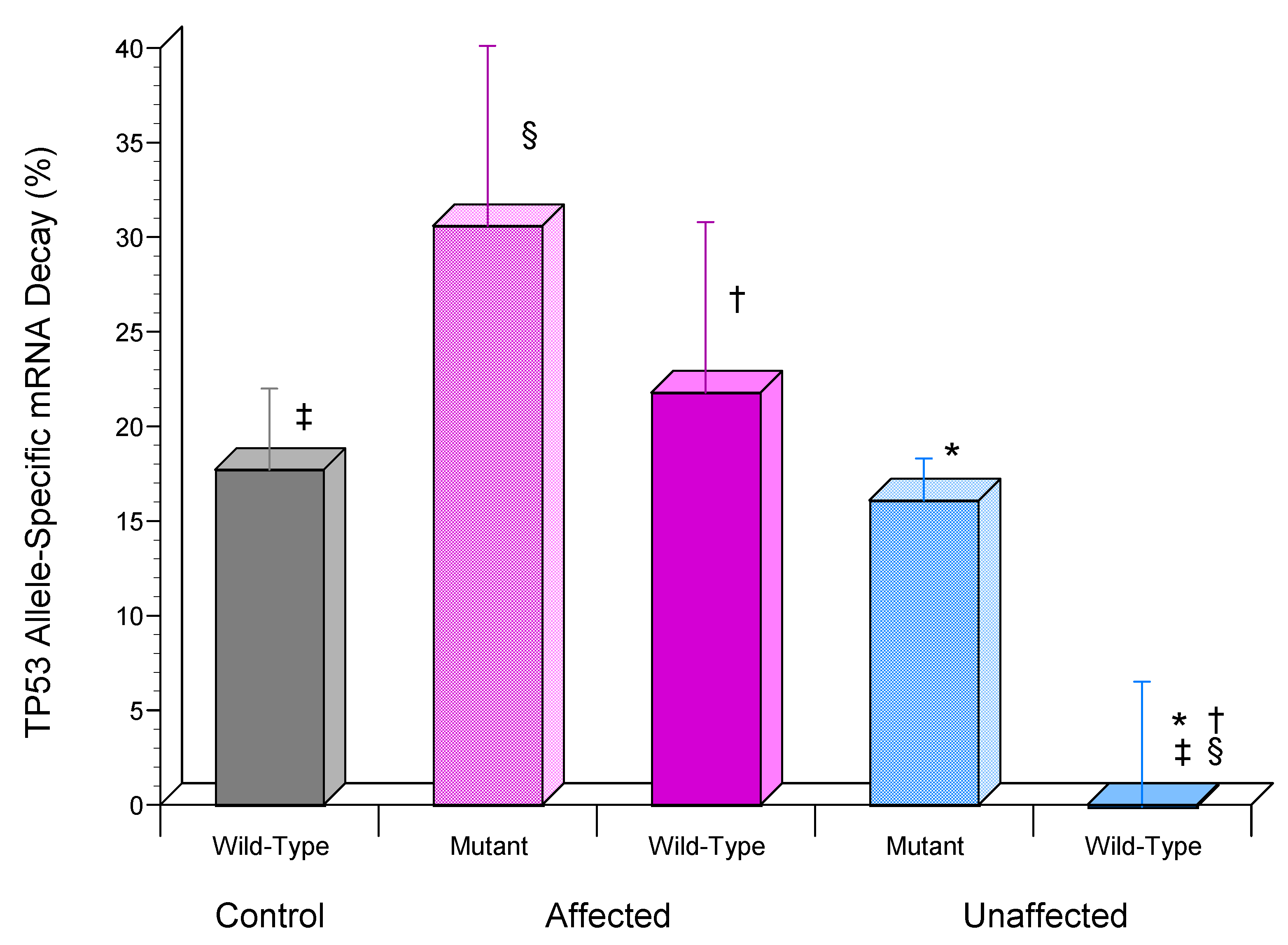
3.5. TP53 mRNA Was Significantly More Stable in Unaffected LFS Fibroblasts
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Höpker, K.; Hagmann, H.; Khurshid, S.; Chen, S.; Schermer, B.; Benzing, T.; Reinhardt, H.C. Putting the brakes on p53-driven apoptosis. Cell Cycle 2012, 11, 4122–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Kamihara, J.; Rana, H.Q.; Garber, J.E. Germline TP53 mutations and the changing landscape of Li-Fraumeni Syndrome. Hum. Mutat. 2014, 35, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Ruijs, M.W.G.; Verhoef, S.; Rookus, M.A.; Pruntel, R.; van der Hout, A.H.; Hogervorst, F.B.L.; Kluijt, I.; Sijmons, R.H.; Aalfs, C.M.; Wagner, A.; et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: Mutation detection rate and relative frequency of cancers in different familial phenotypes. J. Med. Genet. 2010, 47, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villani, A.; Tabori, U.; Schiffman, J.; Shlien, A.; Beyene, J.; Druker, H.; Novokmet, A.; Finlay, J.; Malkin, D. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: A prospective observational study. Lancet Oncol. 2011, 12, 559–567. [Google Scholar] [CrossRef]
- Hoe, K.K.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [Green Version]
- Mai, P.L.; Malkin, D.; Garber, J.E.; Schiffman, J.D.; Weitzel, J.N.; Strong, L.C.; Wyss, O.; Locke, L.; Means, V.; Achatz, M.I.; et al. Li-Fraumeni syndrome: Report of a clinical research workshop and creation of a research consortium. Cancer Genet. 2012, 205, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Toettcher, J.E.; Loewer, A.; Ostheimer, G.J.; Yaffe, M.B.; Tidor, B.; Lahav, G. Distinct mechanisms act in concert to mediate cell cycle arrest. Proc. Natl. Acad. Sci. USA 2009, 106, 785–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey-Boltz, L.A. Bringing It All Together: Coupling Excision Repair to the DNA Damage Checkpoint. Photochem. Photobiol. 2017, 93, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Q.; Carrier, F.; Fornace, A.J., Jr. Induction of cellular p53 activity by DNA-damaging agents and growth arrest. Mol. Cell. Biol. 1993, 13, 4242–4250. [Google Scholar] [CrossRef] [PubMed]
- Bruins, W.; Bruning, O.; Jonker, M.J.; Zwart, E.; van der Hoeven, T.V.; Pennings, J.L.A.; Rauwerda, H.; de Vries, A.; Breit, T.M. The absence of Ser389 phosphorylation in p53 affects the basal gene expression level of many p53-dependent genes and alters the biphasic response to UV exposure in mouse embryonic fibroblasts. Mol. Cell. Biol. 2008, 28, 1974–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Liu, A.; Ming, X.; Deng, P.; Jiang, Y. UV-induced interaction between p38 MAPK and p53 serves as a molecular switch in determining cell fate. FEBS Lett. 2010, 584, 4711–4716. [Google Scholar] [CrossRef] [Green Version]
- Latonen, L.; Taya, Y.; Laiho, M. UV-radiation induces dose-dependent regulation of p53 response and modulates p53-HDM2 interaction in human fibroblasts. Oncogene 2001, 20, 6784–6793. [Google Scholar] [CrossRef] [Green Version]
- Barley, R.D.; Enns, L.; Paterson, M.C.; Mirzayans, R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fibroblast strains heterozygous for TP53 mutations. Oncogene 1998, 17, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Buzby, J.S.; Williams, S.A.; Schaffer, L.; Head, S.R.; Nugent, D.J. Allele-specific wild-type TP53 expression in the unaffected carrier parent of children with Li-Fraumeni syndrome. Cancer Genet. 2017, 211, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, S.J.; Sakaguchi, K.; Lees-Miller, S.P.; Fiscella, M.; Mercer, W.E.; Anderson, C.W.; Appella, E. Phosphorylation at Ser-15 and Ser-392 in mutant p53 molecules from human tumors is altered compared to wild-type p53. Proc. Natl. Acad. Sci. USA 1993, 90, 5954–5958. [Google Scholar] [CrossRef] [Green Version]
- Robles-Espinoza, C.D.; Mohammadi, P.; Bonilla, X.; Gutierrez-Arcelus, M. Allele-specific expression: Applications in cancer and technical considerations. Curr. Opin. Genet. Dev. 2021, 66, 10–19. [Google Scholar] [CrossRef]
- Huschtscha, L.I.; Napier, C.E.; Noble, J.R.; Bower, K.; Au, A.Y.; Campbell, H.G.; Braithwaite, A.W.; Reddel, R.R. Enhanced isolation of fibroblasts from human skin explants. Biotechniques 2012, 53, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, Y.; Chang, M.; Lee, S.M.; Buzby, J.S.; Cairo, M.S. Regulation of interleukin-11 protein and mRNA expression in neonatal and adult fibroblasts and endothelial cells. Blood 1994, 84, 4125–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degasperi, A.; Birtwistle, M.R.; Volinsky, N.; Rauch, J.; Kolch, W.; Kholodenko, B.N. Evaluating strategies to normalise biological replicates of Western blot data. PLoS ONE 2014, 9, e87293. [Google Scholar] [CrossRef] [Green Version]
- Buzby, J.S.; Lee, S.M.; Van Winkle, P.; DeMaria, C.T.; Brewer, G.; Cairo, M.S. Increased granulocyte-macrophage colony-stimulating factor mRNA instability in cord versus adult mononuclear cells is translation-dependent and associated with increased levels of A + U-rich element binding factor. Blood 1996, 88, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- Belasco, J.G.; Brawerman, G. Chapter 18: Experimental approaches to the study of mRNA decay. In Control of Messenger RNA Stability; Belasco, J.G., Brawerman, G., Eds.; Academic Press: Boston, MA, USA, 1993; pp. 476–479. [Google Scholar] [CrossRef]
- Nakashima, H.; Akahoshi, M.; Tanaka, Y. Mutation detection using RT-PCR-RFLP. Methods Mol. Biol. 2003, 226, 319–322. [Google Scholar] [CrossRef]
- Buzby, J.S.; Williams, S.A.; Imfeld, K.L.; Kunicki, T.J.; Nugent, D.J. Tissue factor inflammatory response regulated by promoter genotype and p38 MAPK in neonatal vs. adult microvascular endothelial cells. Inflamm. Res. 2014, 63, 299–308. [Google Scholar] [CrossRef]
- Maréchal, A.; Zhou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- Ünsal-Kaçmaz, K.; Makhov, A.M.; Griffith, J.D.; Sancar, A. Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 6673–6678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.Q.; Stanbridge, E.J.; Lao, X.; Cai, Q.; Fan, S.T.; Redpath, J.L. p53-dependent Chk1 phosphorylation is required for maintenance of prolonged G2 arrest. Radiat. Res. 2007, 168, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.M.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Garritano, S.; Inga, A.; Gemignani, F.; Landi, S. More targets, more pathways and more clues for mutant p53. Oncogenesis 2013, 2, e54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisio, A.; Ciribilli, Y.; Fronza, G.; Inga, A.; Monti, P. TP53 mutants in the tower of babel of cancer progression. Hum. Mutat. 2014, 35, 689–701. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. Regulation of p53 downstream genes. Semin. Cancer Biol. 1998, 8, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plass, C.; Soloway, P.D. DNA methylation, imprinting and cancer. Eur. J. Hum. Genet. 2002, 10, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Saldaña-Meyer, R.; Recillas-Targa, F. Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 2011, 6, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, S.; Martin, E.; Gicquel, C.; Melki, J.; Clark, S.J.; Campbell, P.; Magarey, C.J.; Schulte, K.M.; Röher, H.D.; Delbridge, L.; et al. Mutation and methylation analysis of TP53 in adrenal carcinogenesis. Eur. J. Surg. Oncol. 2005, 31, 549–554. [Google Scholar] [CrossRef]
- Finkova, A.; Vazna, A.; Hrachovina, O.; Bendova, S.; Prochazkova, K.; Sedlacek, Z. The TP53 gene promoter is not methylated in families suggestive of Li-Fraumeni syndrome with no germline TP53 mutations. Cancer Genet. Cytogenet. 2009, 193, 63–66. [Google Scholar] [CrossRef]
- Rosenstierne, M.W.; Vinther, J.; Mittler, G.; Larsen, L.; Mann, M.; Norrild, B. Conserved CPEs in the p53 3’ untranslated region influence mRNA stability and protein synthesis. Anticancer Res. 2008, 28, 2553–2559. [Google Scholar] [PubMed]
- Vilborg, A.; Glahder, J.A.; Wilhelm, M.T.; Bersani, C.; Corcoran, M.; Mahmoudi, S.; Rosenstierne, M.; Grandér, D.; Farnebo, M.; Norrild, B.; et al. The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proc. Natl. Acad. Sci. USA 2009, 106, 15756–15761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucchesi, C.; Zhang, J.; Chen, X. Modulation of the p53 family network by RNA-binding proteins. Transl. Cancer Res. 2016, 5, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Rao, J.N.; Guo, X.; Liu, L.; Santora, R.; Bass, B.L.; Wang, J.Y. Polyamine depletion stabilizes p53 resulting in inhibition of normal intestinal epithelial cell proliferation. Am. J. Physiol. Cell Physiol. 2001, 281, C941–C953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; You, S.; Foster, L.K.; Farris, J.; Foster, D.N. The rapid destabilization of p53 mRNA in immortal chicken embryo fibroblast cells. Oncogene 2001, 20, 5118–5123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fotedar, R.; Bendjennat, M.; Fotedar, A. Role of p21WAF1 in the cellular response to UV. Cell Cycle 2004, 3, 134–137. [Google Scholar] [CrossRef]
- Goto, H.; Kasahara, K.; Inagaki, M. Novel insights into Chk1 regulation by phosphorylation. Cell Struct. Funct. 2015, 40, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.-S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Göhler, T.; Jäger, S.; Warnecke, G.; Yasuda, H.; Kim, E.; Deppert, W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res. 2005, 33, 1087–1100. [Google Scholar] [CrossRef]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buzby, J.S.; Williams, S.A.; Nugent, D.J. Unaffected Li-Fraumeni Syndrome Carrier Parent Demonstrates Allele-Specific mRNA Stabilization of Wild-Type TP53 Compared to Affected Offspring. Genes 2022, 13, 2302. https://doi.org/10.3390/genes13122302
Buzby JS, Williams SA, Nugent DJ. Unaffected Li-Fraumeni Syndrome Carrier Parent Demonstrates Allele-Specific mRNA Stabilization of Wild-Type TP53 Compared to Affected Offspring. Genes. 2022; 13(12):2302. https://doi.org/10.3390/genes13122302
Chicago/Turabian StyleBuzby, Jeffrey S., Shirley A. Williams, and Diane J. Nugent. 2022. "Unaffected Li-Fraumeni Syndrome Carrier Parent Demonstrates Allele-Specific mRNA Stabilization of Wild-Type TP53 Compared to Affected Offspring" Genes 13, no. 12: 2302. https://doi.org/10.3390/genes13122302