
Dynamic Transcriptome Analysis Reveals Transcription Factors Involved in the Synthesis of Ethyl Acetate in Aroma-Producing Yeast

 , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
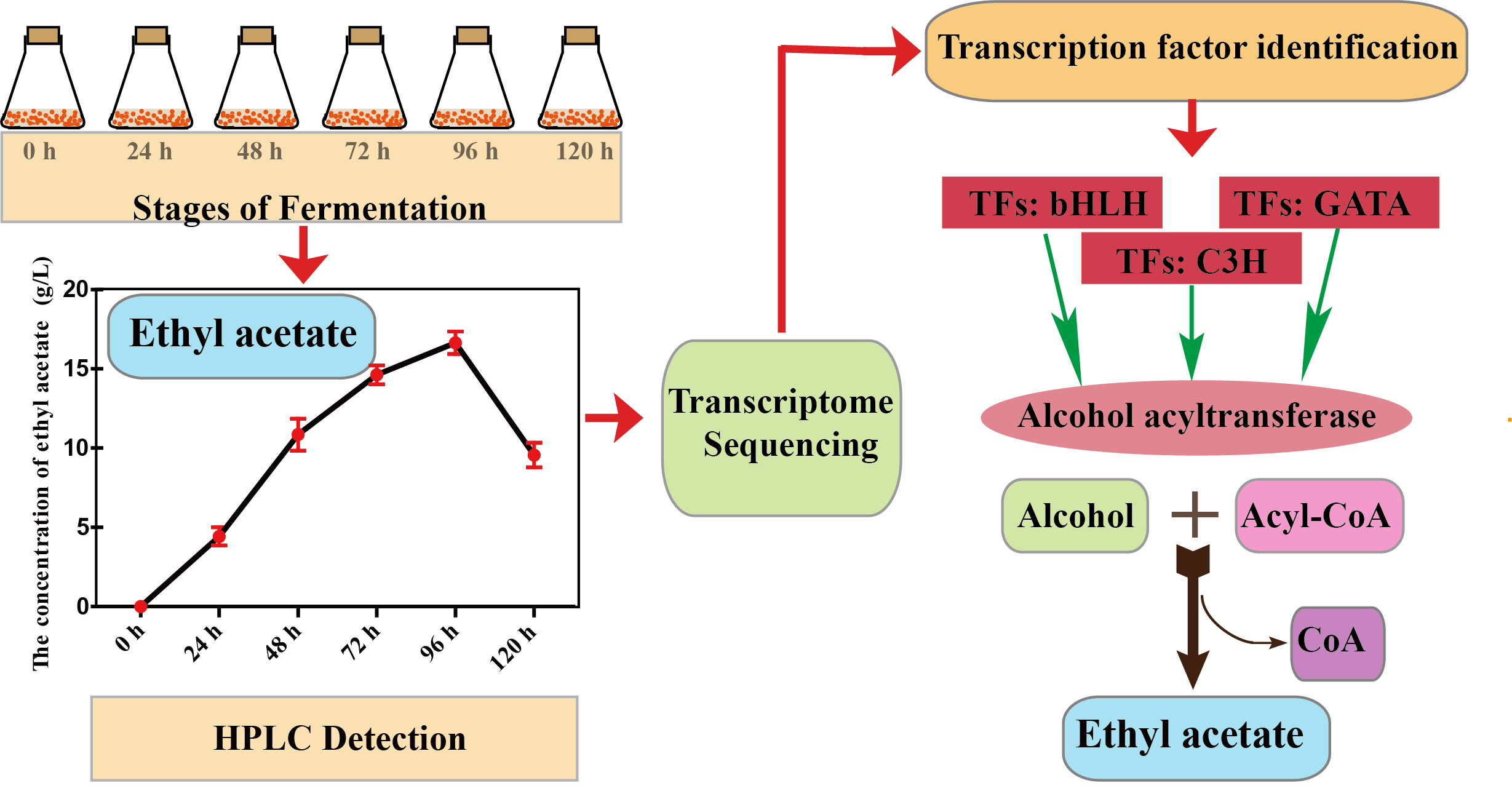
2.1. Sampling
2.2. RNA Extraction and Sequencing
2.3. Transcriptome Sequencing Analysis
2.4. Transcription Factor Identification and Short Time-Series Expression Miner Analysis
2.5. Expression Analysis of Transcription Factors by Real-Time Quantitative PCR
2.6. Statistical Analysis
3. Results
3.1. Overview of the RNA Sequencing Results
3.2. Analysis of DEGs in the Synthesis of Ethyl Acetate
3.3. Identification and Expression Pattern Analysis of Transcription Factors in W. Anomalus Strain YF1503
3.4. Gene Functional Enrichment Analysis of YF1503 Transcription Factors
3.5. Transcription Factors Involved in the Synthesis of Ethyl Acetate
3.6. RT-qPCR Validation of DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Wang, L.; Wang, H.; Yang, F.; Chen, L.; Hao, F.; Lv, X.; Du, H.; Xu, Y. Effects of initial temperature on microbial community succession rate and volatile flavors during Baijiu fermentation process. Food Res. Int. 2021, 141, 109887. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Huang, H.; Pang, Z.; Fu, Z.; Niu, J.; Zhang, C.; Li, W.; Li, X.; Sun, B. Correlation between microbial communities and flavor compounds during the fifth and sixth rounds of sauce-flavor baijiu fermentation. Food Res. Int. 2021, 150, 110741. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Fan, W.; Xu, Y. Characterization of Geosmin as Source of Earthy Odor in Different Aroma Type Chinese Liquors. J Agric. Food Chem. 2011, 59, 8331–8337. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Cao, X.; Cheng, J.; Li, L.; Zhang, T.; Wu, Q.; Xiang, P.; Shen, C.; Li, Q. Chinese Baijiu: The Perfect Works of Microorganisms. Front. Microbiol. 2022, 13, 919044. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhao, D.; Sun, B. Research Progress on the Profile of Trace Components in Baijiu. Food Rev. Int. 2021, 13, 1–27. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, J.; Liu, X.; Zhang, C.; Zhao, Z.; Li, X.; Sun, B. Flavor mystery of Chinese traditional fermented baijiu: The great contribution of ester compounds. Food Chem. 2022, 369, 130920. [Google Scholar] [CrossRef]
- Liu, H.; Sun, B. Effect of fermentation processing on the flavor of Baijiu. J. Agric. Food Chem. 2018, 66, 5425–5432. [Google Scholar] [CrossRef]
- Wei, Y.; Zou, W.; Shen, C.-H.; Yang, J.-G. Basic flavor types and component characteristics of Chinese traditional liquors: A review. J. Food Sci. 2020, 85, 4096–4107. [Google Scholar] [CrossRef]
- Yin, X.; Yoshizaki, Y.; Kurazono, S.; Sugimachi, M.; Takeuchi, H.; Han, X.-L.; Okutsu, K.; Futagami, T.; Tamaki, H.; Takamine, K. Characterization of Flavor Compounds in Rice-flavor baijiu, a Traditional Chinese Distilled Liquor, Compared with Japanese Distilled Liquors, awamori and kome-shochu. Food Sci. Technol. Res. 2020, 26, 411–422. [Google Scholar] [CrossRef]
- Fan, G.; Sun, B.; Dai, X.; Chao, T.; Li, X. Isolation and Identification of High-Yield Ethyl Acetate-Producing Yeast from Gujinggong Daqu and Its Fermentation Characteristics. J. Am. Soc. Brew. Chem. 2018, 76, 117–124. [Google Scholar] [CrossRef]
- Shen, Y.F. Study on the Formation Mechanism of Four Main Kinds of Ethyl Esters in the Fermentation of Liquors. Liquor.-Mak. Sci. Technol. 2003, 5, 28–31. [Google Scholar]
- Zheng, X.-W.; Han, B.-Z. Baijiu (白酒), Chinese liquor: History, classification and manufacture. J. Ethn. Foods 2016, 3, 19–25. [Google Scholar] [CrossRef]
- Chen, Y.; Li, F.; Guo, J.; Liu, G.; Guo, X.; Xiao, D. Enhanced ethyl caproate production of Chinese liquor yeast by overexpressingEHT1with deletedFAA1. J. Ind. Microbiol. Biotechnol. 2014, 41, 563–572. [Google Scholar] [CrossRef]
- Dzialo, M.C.; Park, R.; Steensels, J.; Lievens, B.; Verstrepen, K.J. Physiology, ecology and industrial applications of aroma formation in yeast. FEMS Microbiol. Rev. 2017, 41, S95–S128. [Google Scholar] [CrossRef]
- Van Rijswijck, I.M.H.; Kruis, A.J.; Wolkers–Rooijackers, J.C.M.; Abee, T.; Smid, E.J. Acetate-ester hydrolase activity for screening of the variation in acetate ester yield of Cyberlindnera fabianii, Pichia kudriavzevii and Saccharomyces cerevisiae. LWT 2019, 104, 8–15. [Google Scholar] [CrossRef]
- Kruis, A.J.; Levisson, M.; Mars, A.E.; van der Ploeg, M.; Garcés Daza, F.; Ellena, V.; Kengen, S.W.M.; van der Oost, J.; Weusthuis, R.A. Ethyl acetate production by the elusive alcohol acetyltransferase from yeast. Metab. Eng. 2017, 41, 92–101. [Google Scholar] [CrossRef]
- Ciani, M.; Comitini, F. Yeast interactions in multi-starter wine fermentation. Curr. Opin. Food Sci. 2015, 1, 1–6. [Google Scholar] [CrossRef]
- Wang, X.D.; Ban, S.-D.; Qiu, S.-Y. Analysis of the mould microbiome and exogenous enzyme production in Moutai-flavor Daqu. J. Inst. Brew. 2017, 124, 91–99. [Google Scholar] [CrossRef]
- Zhi-Hui, L.I.; Ren, B.L.; Zhu, J.H.; Ping-Ping, D.U.; Zhang, Y.Y.; Jun-Ping, H.E.; Zhang, W.; Wang, J.; Tan, J.X. Isolation and identification of an ethyl acetate producing yeast strain C-42. Sci. Technol. Food Ind. 2014, 35, 188–191. [Google Scholar]
- Zhilei, F.; Baoguo, S.; Xiuting, L.; Guangsen, F.; Chao, T.; Ahmad, A.; Yingmin, J. Isolation and characterization of a high ethyl acetate-producing yeast from Laobaigan Daqu and its fermentation conditions for producing high-quality Baijiu. Biotechnol. Biotechnol. Equip. 2018, 32, 1218–1227. [Google Scholar]
- Zhang, S.; Guo, F.; Yan, W.; Dong, W.; Zhou, J.; Zhang, W.; Xin, F.; Jiang, M. Perspectives for the microbial production of ethyl acetate. Appl. Microbiol. Biotechnol. 2020, 104, 7239–7245. [Google Scholar] [CrossRef] [PubMed]
- Bigdelou, Z.; Johari, B.; Kadivar, M.; Rismani, E.; Saltanatpour, Z. Investigation of specific binding of designed oligodeoxynucleotide decoys to transcription factors in HT29 cell line undergoing epithelial–mesenchymal transition (EMT). J. Cell. Physiol. 2019, 234, 22765–22774. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Zhang, X.; Zhou, B.; Zhao, K.; Li, R.; Jiang, T. Expression Pattern of ERF Gene Family under Multiple Abiotic Stresses in Populus simonii x P-nigra. Front. Plant Sci. 2017, 8, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Marguerat, S.; Bahler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. CMLS 2010, 67, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, C.; Wan, S.; Zhang, T.; Shan, S. Transcriptomic analysis and discovery of genes in the response of Arachis hypogaea to drought stress. Mol. Biol. Rep. 2018, 45, 119–131. [Google Scholar] [CrossRef]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef]
- Wolf, J.B. Principles of transcriptome analysis and gene expression quantification: An RNA-seq tutorial. Mol. Ecol. Resour. 2013, 13, 559–572. [Google Scholar] [CrossRef]
- Wang, Z.X.; Zhou, X.-Z.; Meng, H.-M.; Liu, Y.-J.; Zhou, Q.; Huang, B. Comparative transcriptomic analysis of the heat stress response in the filamentous fungusMetarhizium anisopliaeusing RNA-Seq. Appl. Microbiol. Biotechnol. 2014, 98, 5589–5597. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Yao, W.; Zhou, B.; Zhang, X.; Zhao, K.; Cheng, Z.; Jiang, T. Transcriptome analysis of transcription factor genes under multiple abiotic stresses in Populus simonii × P. nigra. Gene 2019, 707, 189–197. [Google Scholar] [CrossRef]
- Wan, X.L.; Qiao, Z.; Yuan, W.Y.; En, W.W.; Zhu, B.M.; Wei, Z.J. Identification of heat-responsive genes in carnation (Dianthus caryophyllus L.) by RNA-seq. Front. Plant Sci. 2015, 6, 519. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zhang, J.; Tian, X.; Wu, S.; Zhang, Q.; Zhang, J.; Dang, Z.; Pei, X. De novo assembly of the desert tree Haloxylon ammodendron (C. A. Mey.) based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 1111. [Google Scholar] [CrossRef] [PubMed]
- Parafati, L.; Cirvilleri, G.; Restuccia, C.; Wisniewski, M. Potential Role of Exoglucanase Genes (WaEXG1 and WaEXG2) in the Biocontrol Activity of Wickerhamomyces anomalus. Microb. Ecol. 2017, 73, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Kurita, O. Increase of acetate ester-hydrolysing esterase activity in mixed cultures of Saccharomyces cerevisiae and Pichia anomala. J. Appl. Microbiol. 2008, 104, 1051–1058. [Google Scholar] [CrossRef]
- Yong, C.P.; Shaffer, C.E.H.; Bennett, G.N. Microbial formation of esters. Appl. Microbiol. Biotechnol. 2009, 85, 13–25. [Google Scholar]
- Loeser, C.; Urit, T.; Bley, T. Perspectives for the biotechnological production of ethyl acetate by yeasts. Appl. Microbiol. Biotechnol. 2014, 98, 5397–5415. [Google Scholar] [CrossRef]
- Hu, X.; Tian, R.; Wang, K.; Cao, Z.; Yan, P.; Li, F.; Li, X.; Li, S.; He, P. The prokaryotic community, physicochemical properties and flavors dynamics and their correlations in fermented grains for Chinese strong-flavor Baijiu production. Food Res. Int. 2021, 148, 110626. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Y.; Huang, Y. Microbiome diversity and evolution in stacking fermentation during different rounds of Jiang-flavoured Baijiu brewing. LWT 2021, 143, 111119. [Google Scholar] [CrossRef]
- Ling, X.U.; Zhang, Q.Y. Screening and Application of Several Aroma-producing Yeast Strains. Liquor.-Mak. Sci. Technol. 2010, 12, 48–54. [Google Scholar]
- Xu, Y.; Sun, B.; Fan, G.; Teng, C.; Xiong, K.; Zhu, Y.; Li, J.; Li, X. The brewing process and microbial diversity of strong flavour Chinese spirits: A review. J. Inst. Brew. 2017, 123, 5–12. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Data Rate (%) | Q20(%) | GC Content(%) |
|---|---|---|---|---|---|
| 0 h | 46,040,584 | 45,382,701 | 98.57 | 97.91 | 37.19 |
| 24 h | 44,570,516 | 43,779,746 | 98.23 | 97.94 | 39.17 |
| 48 h | 44,851,995 | 44,058,827 | 98.23 | 98.03 | 37.81 |
| 72 h | 43,271,427 | 41,956,821 | 96.96 | 97.96 | 37.85 |
| 96 h | 45,610,435 | 44,290,539 | 97.11 | 97.92 | 42.22 |
| 120 h | 43,967,872 | 42,710,191 | 97.14 | 97.93 | 38.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, B.; Li, W.; Ifrah, K.; Du, B.; Xu, Y.; Zhang, C.; Li, X. Dynamic Transcriptome Analysis Reveals Transcription Factors Involved in the Synthesis of Ethyl Acetate in Aroma-Producing Yeast. Genes 2022, 13, 2341. https://doi.org/10.3390/genes13122341
Ni B, Li W, Ifrah K, Du B, Xu Y, Zhang C, Li X. Dynamic Transcriptome Analysis Reveals Transcription Factors Involved in the Synthesis of Ethyl Acetate in Aroma-Producing Yeast. Genes. 2022; 13(12):2341. https://doi.org/10.3390/genes13122341
Chicago/Turabian StyleNi, Bingqian, Weiwei Li, Kiren Ifrah, Binghao Du, Youqiang Xu, Chengnan Zhang, and Xiuting Li. 2022. "Dynamic Transcriptome Analysis Reveals Transcription Factors Involved in the Synthesis of Ethyl Acetate in Aroma-Producing Yeast" Genes 13, no. 12: 2341. https://doi.org/10.3390/genes13122341
APA StyleNi, B., Li, W., Ifrah, K., Du, B., Xu, Y., Zhang, C., & Li, X. (2022). Dynamic Transcriptome Analysis Reveals Transcription Factors Involved in the Synthesis of Ethyl Acetate in Aroma-Producing Yeast. Genes, 13(12), 2341. https://doi.org/10.3390/genes13122341