




A Framework for Comparison and Assessment of Synthetic RNA-Seq Data

Abstract
:1. Introduction
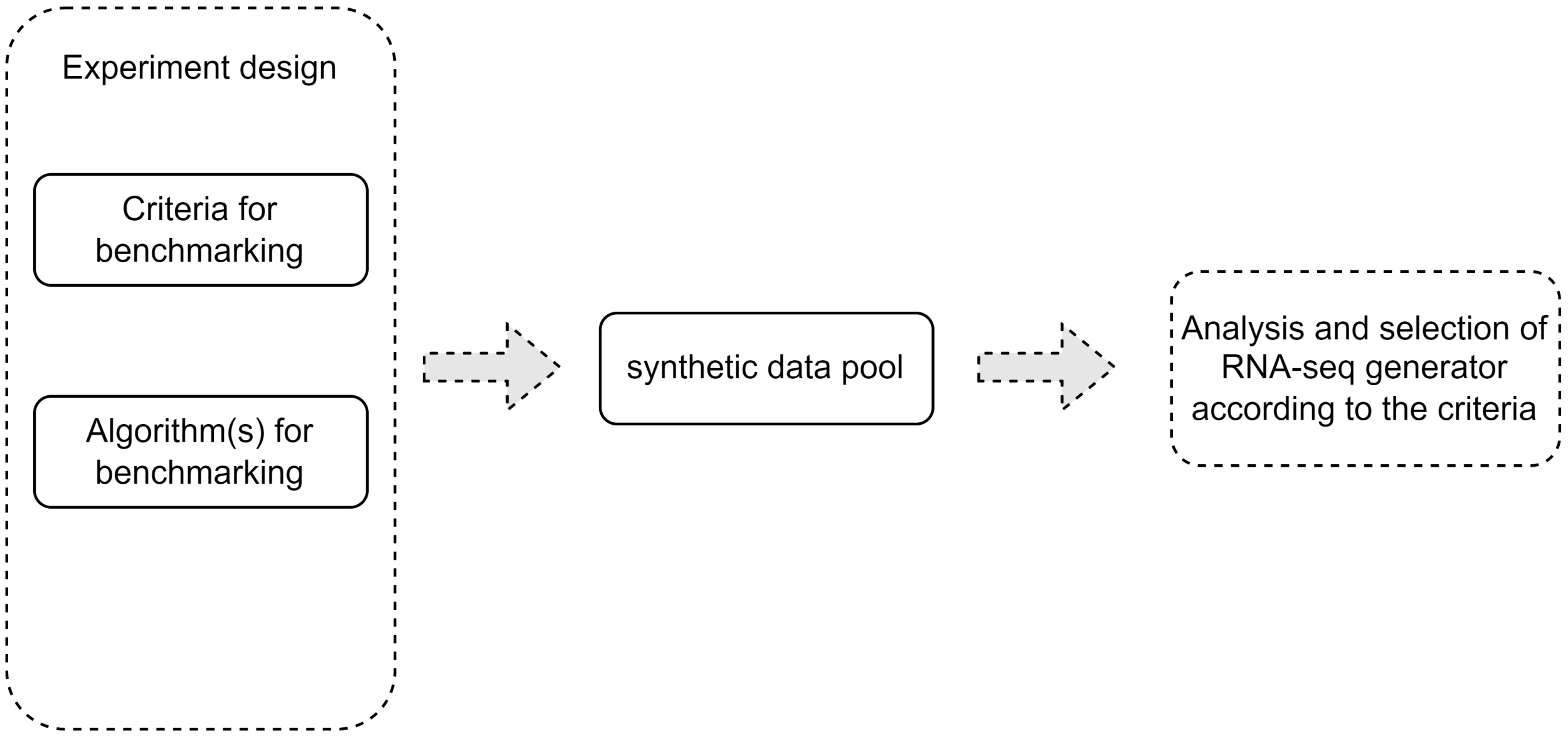
2. A General Computational Framework for Selection of Task-Specific Synthetic RNA-Seq Data Generator
3. Application of the Framework to Benchmark Several Synthetic Bulk RNA-Seq Data Generators
- (i)
- We calculated the parameters needed for the benchmarked packages using the NGSSPPG synthetic RNA-seq datasets or the real RNA-seq data (AD dataset) as input for textbfcompcodeR: mean gene expression of class 1, and specific dispersions of class 1 and class 2, as recommended in the package manual. We used the default values for effect size and the minfact and maxfact parameters for the simulated samples’ individual sequencing depths.
- (ii)
- powsimR’s parameter estimation step was performed with the recommended settings for bulk RNA-seq data; the simulation step was performed with the DESeq2 differential testing method. For this particular simulation, we truncated the values above to be equal to for the AD dataset, with the goal of avoiding the effect of severe outliers.
- (iii)
- As the input of seqgendiff, we took either one of the two classes from the NGSSPPG dataset or the control group from the AD dataset. The added signal is from an exponential distribution with a rate of 0.5 and effect size of 1.5.
- (iv)
- SimSeq runs as a one-step procedure; therefore, we used its default parameters.
- (v)
- For SPsimSeq, we set the genewiseCor parameter to FALSE; therefore, we chose not to retain the gene-to-gene correlation structure of the input data. This was to avoid the high computational cost of calculating/keeping that structure.
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DEGs | Differentially expressed genes |
| RNA-seq | RNA-sequencing |
| scRNA-seq | single cell RNA-sequencing |
References
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2010, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Thind, A.S.; Monga, I.; Thakur, P.K.; Kumari, P.; Dindhoria, K.; Krzak, M.; Ranson, M.; Ashford, B. Demystifying emerging bulk RNA-Seq applications: The application and utility of bioinformatic methodology. Brief. Bioinform. 2021, 22, bbab259. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Clevers, H. Coexistence of Quiescent and Active Adult Stem Cells in Mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S. Non-genetic heterogeneity of cells in development: More than just noise. Development 2009, 136, 3853–3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberwine, J.; Yeh, H.; Miyashiro, K.; Cao, Y.; Nair, S.; Finnell, R.; Zettel, M.; Coleman, P. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA 1992, 89, 3010–3014. [Google Scholar] [CrossRef] [Green Version]
- Brady, G.; Barbara, M.; Iscove, N.N. Representative in vitro cDNA amplification from individual hemopoietic cells and colonies. Methods Mol. Cell Biol. 1990, 2, 17–25. [Google Scholar]
- Klein, C.A.; Seidl, S.; Petat-Dutter, K.; Offner, S.; Geigl, J.B.; Schmidt-Kittler, O.; Wendler, N.; Passlick, B.; Huber, R.M.; Schlimok, G.; et al. Combined transcriptome and genome analysis of single micrometastatic cells. Nat. Biotechnol. 2002, 20, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Tang, L. Single-cell profiling of microbes. Nat. Methods 2021, 18, 334. [Google Scholar] [CrossRef] [PubMed]
- Hegenbarth, J.C.; Lezzoche, G.; Windt, L.J.D.; Stoll, M. Perspectives on Bulk-Tissue RNA Sequencing and Single-Cell RNA Sequencing for Cardiac Transcriptomics. Front. Mol. Med. 2022, 2, 839338. [Google Scholar] [CrossRef]
- Svensson, V.; Vento-Tormo, R.; Teichmann, S.A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018, 13, 599–604. [Google Scholar] [CrossRef]
- Jiang, R.; Sun, T.; Song, D.; Li, J.J. Statistics or biology: The zero-inflation controversy about scRNA-seq data. Genome Biol. 2022, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Rai, A.; Merchant, M.L.; Cave, M.C.; Rai, S.N. A Comprehensive Survey of Statistical Approaches for Differential Expression Analysis in Single-Cell RNA Sequencing Studies. Genes 2021, 12, 1947. [Google Scholar] [CrossRef]
- Bijl, B. How Much Does Single-Cell Sequencing Cost? Available online: https://www.scdiscoveries.com/blog/knowledge/cost-of-single-cell-sequencing/ (accessed on 11 July 2022).
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. scmap: Projection of single-cell RNA-seq data across datasets. Nat. Methods 2018, 15, 359–362. [Google Scholar] [CrossRef]
- Abugessaisa, I.; Noguchi, S.; Böttcher, M.; Hasegawa, A.; Kouno, T.; Kato, S.; Tada, Y.; Ura, H.; Abe, K.; Shin, J.W.; et al. SCPortalen: Human and mouse single-cell centric database. Nucleic Acids Res. 2017, 46, D781–D787. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhu, J.; Han, G.; Jia, P.; Zhao, Z. scRNASeqDB: A database for gene expression profiling in human single cell by RNA-seq. Genes 2017, 8, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zheng, X.; Wang, J.; Wan, S.; Song, F.; Wong, M.H.; Leung, K.S.; Cheng, L. Improving bulk RNA-seq classification by transferring gene signature from single cells in acute myeloid leukemia. Brief. Bioinform. 2022, 23, bbac002. [Google Scholar] [CrossRef]
- Das, S.; Rai, A.; Mishra, D.; Rai, S.N. Statistical approach for selection of biologically informative genes. Gene 2018, 655, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Rai, S.N. Statistical Approach for Biologically Relevant Gene Selection from High-Throughput Gene Expression Data. Entropy 2020, 22, 1205. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Rai, A.; Rai, S.N. Differential Expression Analysis of Single-Cell RNA-Seq Data: Current Statistical Approaches and Outstanding Challenges. Entropy 2022, 24, 995. [Google Scholar] [CrossRef] [PubMed]
- Arowolo, M.O.; Adebiyi, M.O.; Aremu, C.; Adebiyi, A.A. A survey of dimension reduction and classification methods for RNA-Seq data on malaria vector. J. Big Data 2021, 8, 50. [Google Scholar] [CrossRef]
- Johnson, N.T.; Dhroso, A.; Hughes, K.J.; Korkin, D. Biological classification with RNA-seq data: Can alternatively spliced transcript expression enhance machine learning classifiers? RNA 2018, 24, 1119–1132. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, R. Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods 2013, 11, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sham, P.; Tong, T.; Pang, H. Pathway-Based Single-Cell RNA-Seq Classification, Clustering, and Construction of Gene-Gene Interactions Networks Using Random Forests. IEEE J. Biomed. Health Inform. 2020, 24, 1814–1822. [Google Scholar] [CrossRef]
- Menon, V. Clustering single cells: A review of approaches on high-and low-depth single-cell RNA-seq data. Brief. Funct. Genom. 2017, 17, 240–245. [Google Scholar] [CrossRef]
- Hu, J.; Li, X.; Hu, G.; Lyu, Y.; Susztak, K.; Li, M. Iterative transfer learning with neural network for clustering and cell type classification in single-cell RNA-seq analysis. Nat. Mach. Intell. 2020, 2, 607–618. [Google Scholar] [CrossRef]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A Gene-Coexpression Network for Global Discovery of Conserved Genetic Modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.M.; Fabbro, C.D.; Licausi, F. Comparative study of RNA-seq- and Microarray-derived coexpression networks in Arabidopsis thaliana. Bioinformatics 2013, 29, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Ballouz, S.; Verleyen, W.; Gillis, J. Guidance for RNA-seq co-expression network construction and analysis: Safety in numbers. Bioinformatics 2015, 31, 2123–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform. Biol. Insights 2015, 9s1, BBI.S28991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmatallah, Y.; Emmert-Streib, F.; Glazko, G. Comparative evaluation of gene set analysis approaches for RNA-Seq data. BMC Bioinform. 2014, 15, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.E.; Stumpf, M.P.; Babtie, A.C. Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures. Cell Syst. 2017, 5, 251–267.e3. [Google Scholar] [CrossRef] [Green Version]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Kartha, V.K.; Duarte, F.M.; Hu, Y.; Ma, S.; Chew, J.G.; Lareau, C.A.; Earl, A.; Burkett, Z.D.; Kohlway, A.S.; Lebofsky, R.; et al. Functional inference of gene regulation using single-cell multi-omics. Cell Genom. 2022, 2, 100166. [Google Scholar] [CrossRef]
- Soneson, C. compcodeR—An R package for benchmarking differential expression methods for RNA-seq data. Bioinformatics 2014, 30, 2517–2518. [Google Scholar] [CrossRef] [Green Version]
- Zararsız, G.; Goksuluk, D.; Korkmaz, S.; Eldem, V.; Zararsiz, G.E.; Duru, I.P.; Ozturk, A. A comprehensive simulation study on classification of RNA-Seq data. PLoS ONE 2017, 12, e0182507. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, R.; Reiss, D.J.; Shannon, P.; Facciotti, M.; Hood, L.; Baliga, N.S.; Thorsson, V. The Inferelator: An algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006, 7, R36. [Google Scholar] [CrossRef] [PubMed]
- Lasri, A.; Shahrezaei, V.; Sturrock, M. Benchmarking imputation methods for network inference using a novel method of synthetic scRNA-seq data generation. BMC Bioinform. 2022, 23, 236. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigaill, G.; Balzergue, S.; Brunaud, V.; Blondet, E.; Rau, A.; Rogier, O.; Caius, J.; Maugis-Rabusseau, C.; Soubigou-Taconnat, L.; Aubourg, S.; et al. Synthetic data sets for the identification of key ingredients for RNA-seq differential analysis. Brief. Bioinform. 2016, 19, bbw092. [Google Scholar] [CrossRef]
- Esnaola, M.; Puig, P.; Gonzalez, D.; Castelo, R.; Gonzalez, J.R. A flexible count data model to fit the wide diversity of expression profiles arising from extensively replicated RNA-seq experiments. BMC Bioinform. 2013, 14, 254. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.H.; Xia, K.; Wright, F.A. A powerful and flexible approach to the analysis of RNA sequence count data. Bioinformatics 2011, 27, 2672–2678. [Google Scholar] [CrossRef] [Green Version]
- Deaton, A.M.; Webb, S.; Kerr, A.R.; Illingworth, R.S.; Guy, J.; Andrews, R.; Bird, A. Cell type–specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011, 21, 1074–1086. [Google Scholar] [CrossRef] [Green Version]
- Vieth, B.; Ziegenhain, C.; Parekh, S.; Enard, W.; Hellmann, I. powsimR: Power analysis for bulk and single cell RNA-seq experiments. Bioinformatics 2017, 33, 3486–3488. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D.; et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018, 174, 716–729.e27. [Google Scholar] [CrossRef] [Green Version]
- Li, W.V.; Li, J.J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Korthauer, K.D.; Chu, L.F.; Newton, M.A.; Li, Y.; Thomson, J.; Stewart, R.; Kendziorski, C. A statistical approach for identifying differential distributions in single-cell RNA-seq experiments. Genome Biol. 2016, 17, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappia, L.; Phipson, B.; Oshlack, A. Splatter: Simulation of single-cell RNA sequencing data. Genome Biol. 2017, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.; Perraudeau, F.; Gribkova, S.; Dudoit, S.; Vert, J.P. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun. 2018, 9, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Berge, K.V.; Perraudeau, F.; Soneson, C.; Love, M.I.; Risso, D.; Vert, J.P.; Robinson, M.D.; Dudoit, S.; Clement, L. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 2018, 19, 24. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Yang, P.; Yang, J.Y.H. A benchmark study of simulation methods for single-cell RNA sequencing data. Nat. Commun. 2021, 12, 6911. [Google Scholar] [CrossRef]
- Huang, M.; Ye, X.; Li, H.; Sakurai, T. Missing Value Imputation With Low-Rank Matrix Completion in Single-Cell RNA-Seq Data by Considering Cell Heterogeneity. Front. Genet. 2022, 13, 952649. [Google Scholar] [CrossRef]
- Malec, M.; Kurban, H.; Dalkilic, M. ccImpute: An accurate and scalable consensus clustering based algorithm to impute dropout events in the single-cell RNA-seq data. BMC Bioinform. 2022, 23, 291. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, X. BASS: Multi-scale and multi-sample analysis enables accurate cell type clustering and spatial domain detection in spatial transcriptomic studies. Genome Biol. 2022, 23, 168. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Z.; Bhadani, R.; Cao, S.; Lu, M.; Lytal, N.; Chen, Y.; An, L. NISC: Neural Network-Imputation for Single-Cell RNA Sequencing and Cell Type Clustering. Front. Genet. 2022, 13, 847112. [Google Scholar] [CrossRef]
- Zubair, A.; Chapple, R.H.; Natarajan, S.; Wright, W.C.; Pan, M.; Lee, H.M.; Tillman, H.; Easton, J.; Geeleher, P. Cell type identification in spatial transcriptomics data can be improved by leveraging cell-type-informative paired tissue images using a Bayesian probabilistic model. Nucleic Acids Res. 2022, 50, e80. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, P.; Ray, S. A Regularized Multi-Task Learning Approach for Cell Type Detection in Single-Cell RNA Sequencing Data. Front. Genet. 2022, 13, 788832. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, D.; Qu, H. Systematic review of next-generation sequencing simulators: Computational tools, features and perspectives. Brief. Funct. Genom. 2016, 16, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakola, F.; Palejev, D.; Ivanov, I. Comparative Study of Synthetic Bulk RNA-Seq Generators. In Bioinformatics and Biomedical Engineering; Springer International Publishing: Cham, Switzerland, 2022; pp. 57–70. [Google Scholar] [CrossRef]
- Riquier, S.; Bessiere, C.; Guibert, B.; Bouge, A.L.; Boureux, A.; Ruffle, F.; Audoux, J.; Gilbert, N.; Xue, H.; Gautheret, D.; et al. Kmerator Suite: Design of specific k-mer signatures and automatic metadata discovery in large RNA-seq datasets. NAR Genom. Bioinform. 2021, 3, lqab058. [Google Scholar] [CrossRef]
- Alaimo, S.; Maria, A.D.; Shasha, D.; Ferro, A.; Pulvirenti, A. TACITuS: Transcriptomic data collector, integrator, and selector on big data platform. BMC Bioinform. 2019, 20, 366. [Google Scholar] [CrossRef] [Green Version]
- Karathanasis, N.; Tsamardinos, I.; Lagani, V. omicsNPC: Applying the Non-Parametric Combination Methodology to the Integrative Analysis of Heterogeneous Omics Data. PLoS ONE 2016, 11, e0165545. [Google Scholar] [CrossRef] [Green Version]
- Hawinkel, S.; Bijnens, L.; Cao, K.A.L.; Thas, O. Model-based joint visualization of multiple compositional omics datasets. NAR Genom. Bioinform. 2020, 2, lqaa050. [Google Scholar] [CrossRef]
- Klingenberg, H.; Meinicke, P. How to normalize metatranscriptomic count data for differential expression analysis. PeerJ 2017, 5, e3859. [Google Scholar] [CrossRef] [Green Version]
- Lewitus, E.; Rolland, M. A non-parametric analytic framework for within-host viral phylogenies and a test for HIV-1 founder multiplicity. Virus Evol. 2019, 5, vez044. [Google Scholar] [CrossRef]
- Knyazev, S.; Tsyvina, V.; Shankar, A.; Melnyk, A.; Artyomenko, A.; Malygina, T.; Porozov, Y.B.; Campbell, E.M.; Switzer, W.M.; Skums, P.; et al. Accurate assembly of minority viral haplotypes from next-generation sequencing through efficient noise reduction. Nucleic Acids Res. 2021, 49, e102. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, A.; Knyazev, S.; Vannberg, F.; Bunimovich, L.; Skums, P.; Zelikovsky, A. Using earth mover’s distance for viral outbreak investigations. BMC Genom. 2020, 21, 582. [Google Scholar] [CrossRef] [PubMed]
- Gerard, D. Data-based RNA-seq simulations by binomial thinning. BMC Bioinform. 2020, 21, 206. [Google Scholar] [CrossRef] [PubMed]
- Benidt, S.; Nettleton, D. SimSeq: A nonparametric approach to simulation of RNA-sequence datasets. Bioinformatics 2015, 31, 2131–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assefa, A.T.; Vandesompele, J.; Thas, O. SPsimSeq: Semi-parametric simulation of bulk and single-cell RNA-sequencing data. Bioinformatics 2020, 36, 3276–3278. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef]
- Wilks, C.; Zheng, S.C.; Chen, F.Y.; Charles, R.; Solomon, B.; Ling, J.P.; Imada, E.L.; Zhang, D.; Joseph, L.; Leek, J.T.; et al. recount3: Summaries and queries for large-scale RNA-seq expression and splicing. Genome Biol. 2021, 22, 323. [Google Scholar] [CrossRef]
- Dougherty, E.; Hua, J.; Sima, C. Performance of Feature Selection Methods. Curr. Genom. 2009, 10, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, N.; Yousefi, M.R.; Johnson, C.D.; Ivanov, I.; Dougherty, E.R. Modeling the next generation sequencing sample processing pipeline for the purposes of classification. BMC Bioinform. 2013, 14, 307. [Google Scholar] [CrossRef] [Green Version]
- Wilk, M.B.; Gnanadesikan, R. Probability plotting methods for the analysis for the analysis of data. Biometrika 1968, 55, 1–17. [Google Scholar] [CrossRef]
- Soneson, C.; Robinson, M.D. Towards unified quality verification of synthetic count data with countsimQC. Bioinformatics 2017, 34, 691–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermaid, A.; Monier, B.; Zhao, J.; Liu, B.; Ma, Q. Interpretation of differential gene expression results of RNA-seq data: Review and integration. Briefs. Boinform. 2018, 20, 2044–2054. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | # DEGs | Dataset | # DEGs | Dataset | # DEGs |
|---|---|---|---|---|---|
| NGSSPPG1 * | 623 | NGSSPPG2 * | 677 | AD * | 10066 |
| compcodeR | 560 | compcodeR | 453 | compcodeR | 1689 |
| powsimR | 543 | powsimR | 449 | powsimR | 1751 |
| seqgendiff | 573 | seqgendiff | 648 | seqgendiff | 1649 |
| simseq | 77 | simseq | 238 | simseq | 297 |
| SPsimSeq | 629 | SPsimSeq | 674 | SPsimSeq | 1828 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakola, F.; Palejev, D.; Ivanov, I. A Framework for Comparison and Assessment of Synthetic RNA-Seq Data. Genes 2022, 13, 2362. https://doi.org/10.3390/genes13122362
Shakola F, Palejev D, Ivanov I. A Framework for Comparison and Assessment of Synthetic RNA-Seq Data. Genes. 2022; 13(12):2362. https://doi.org/10.3390/genes13122362
Chicago/Turabian StyleShakola, Felitsiya, Dean Palejev, and Ivan Ivanov. 2022. "A Framework for Comparison and Assessment of Synthetic RNA-Seq Data" Genes 13, no. 12: 2362. https://doi.org/10.3390/genes13122362
APA StyleShakola, F., Palejev, D., & Ivanov, I. (2022). A Framework for Comparison and Assessment of Synthetic RNA-Seq Data. Genes, 13(12), 2362. https://doi.org/10.3390/genes13122362