
A Comparative Transcriptomic with UPLC-Q-Exactive MS Reveals Differences in Gene Expression and Components of Iridoid Biosynthesis in Various Parts of Gentiana macrophylla
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and Sequencing
2.3. Transcript Assembly and Unigene Functional Annotation
2.4. Analysis of Differentially Expressed Genes
2.5. Identification of Transcription Factors
2.6. qRT-PCR Validation of Key Genes in the Biosynthesis of Iridoid Compounds
2.7. Analysis of Constituents by UPLC-Q-Exactive MS
2.7.1. Sample Preparation
2.7.2. UPLC-Q-Exactive MS Analysis
2.7.3. UPLC-Q-Exactive MS Data Acquisition and Analysis
2.8. Correlation Analysis between Expression of Key Enzyme Genes and Constituents
2.9. Statistical Analysis
3. Results
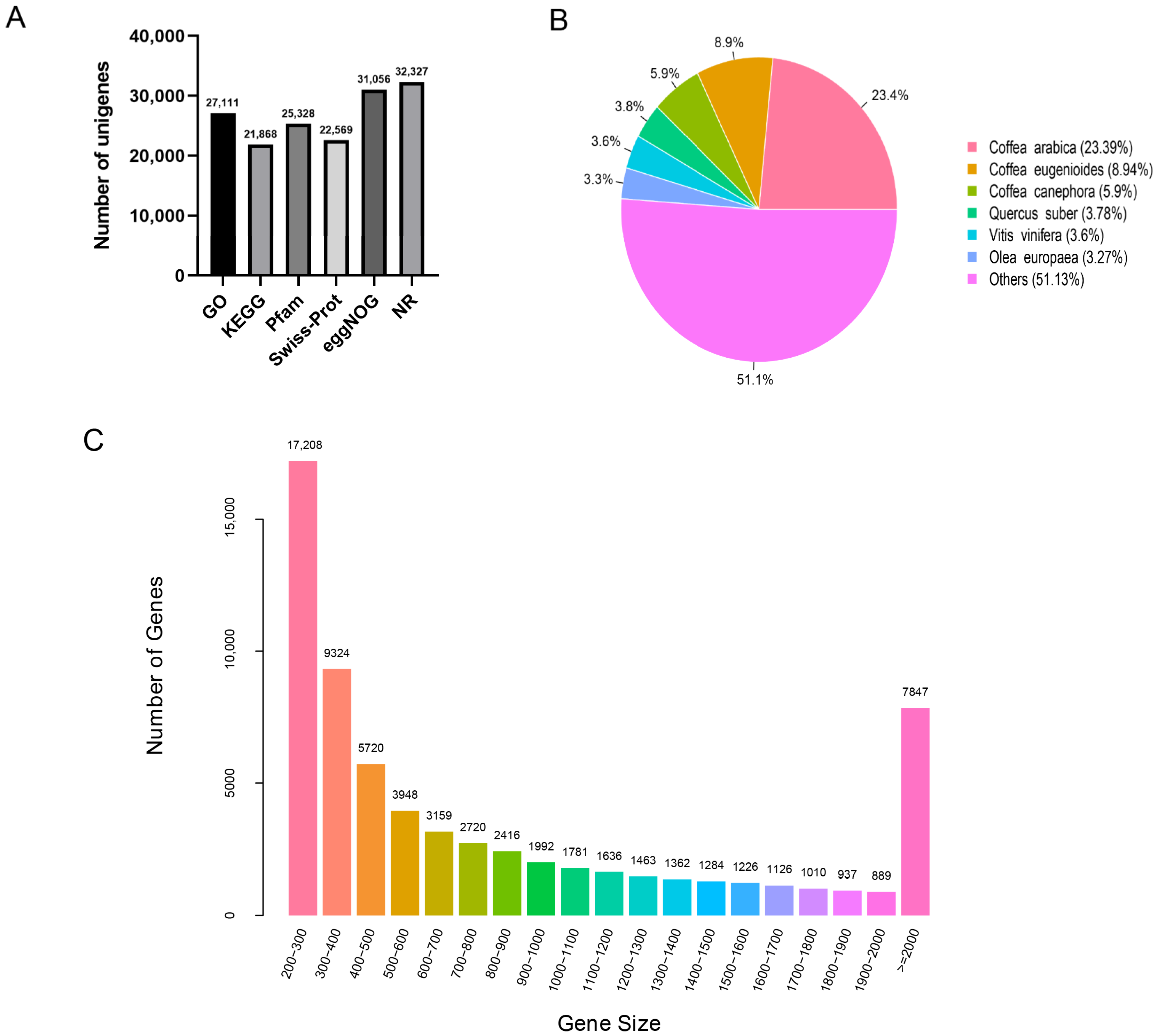
3.1. RNA-Seq and De Novo Transcriptome Assembly
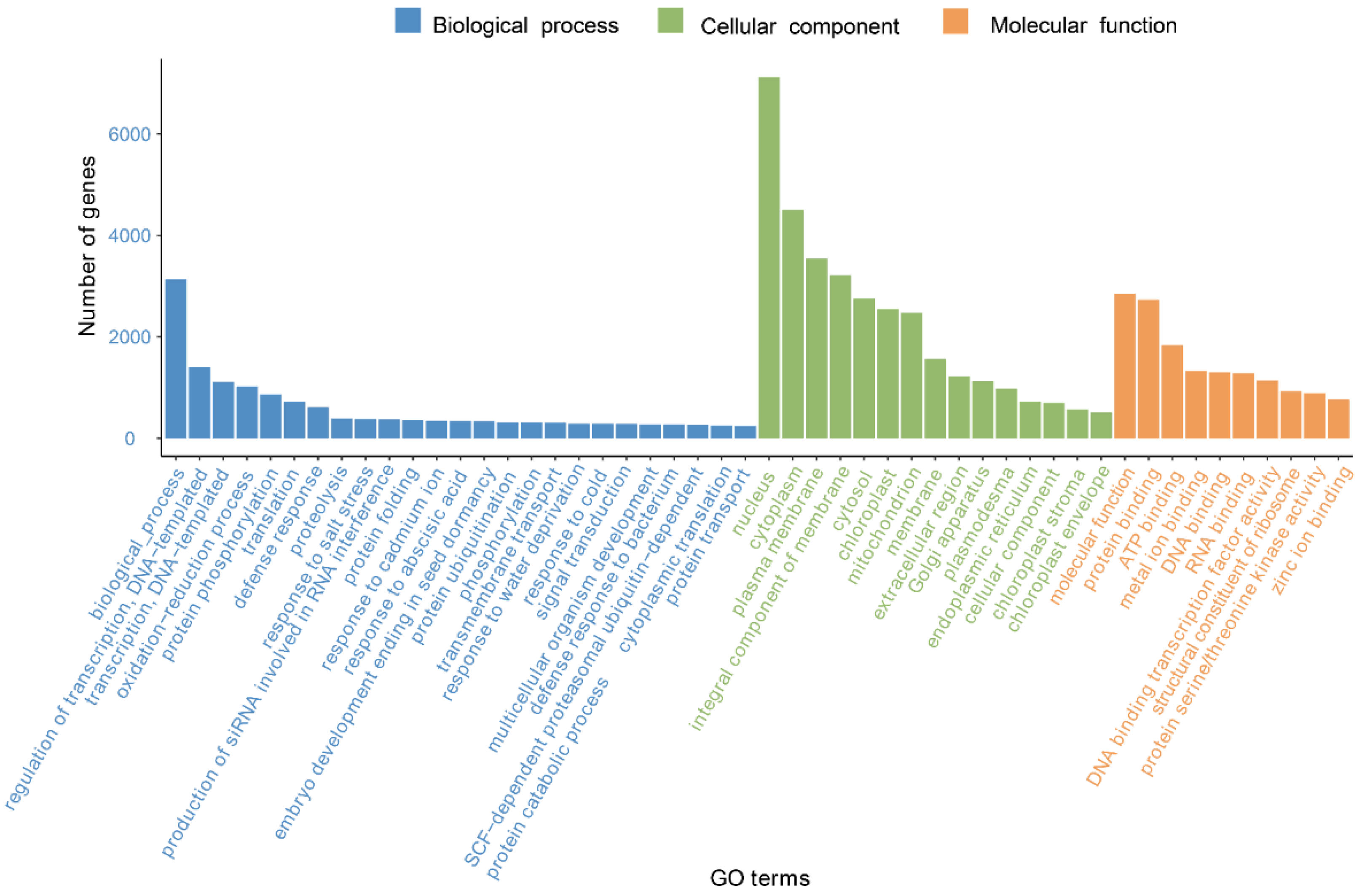
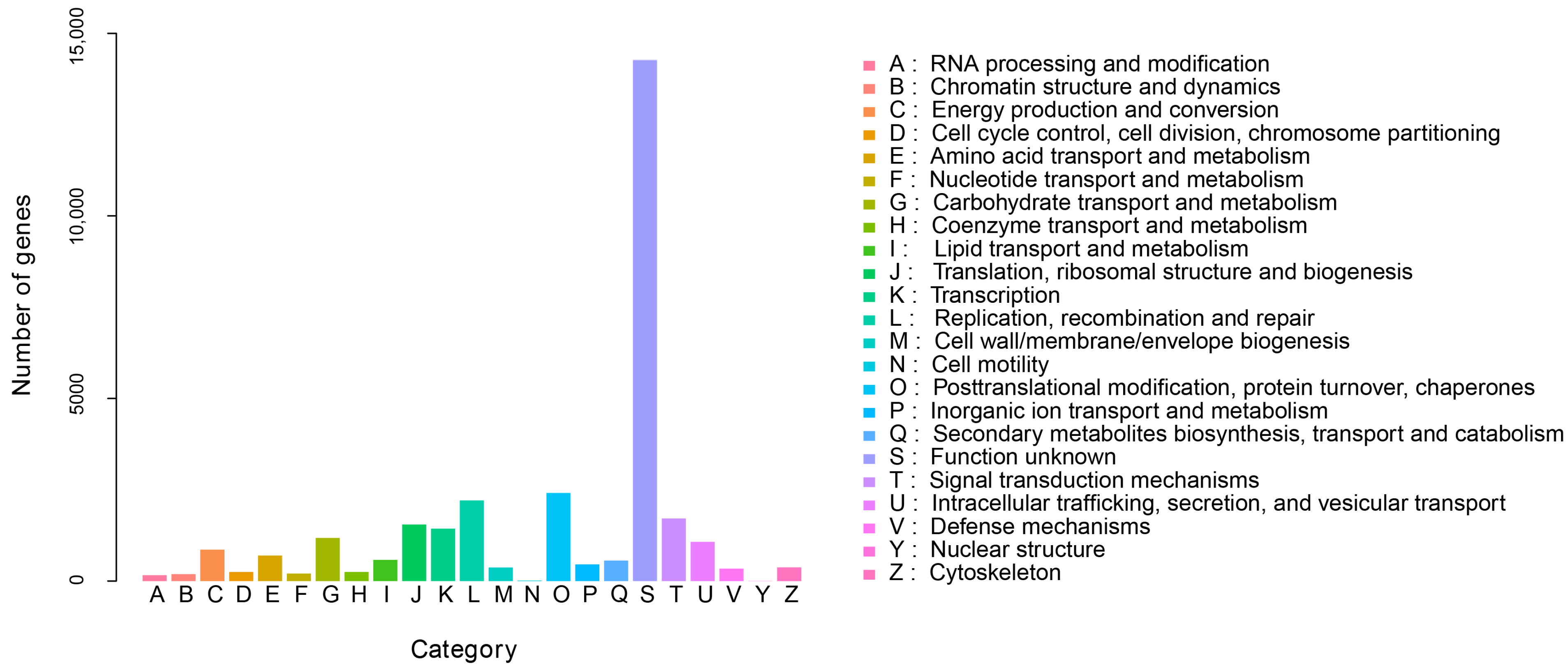
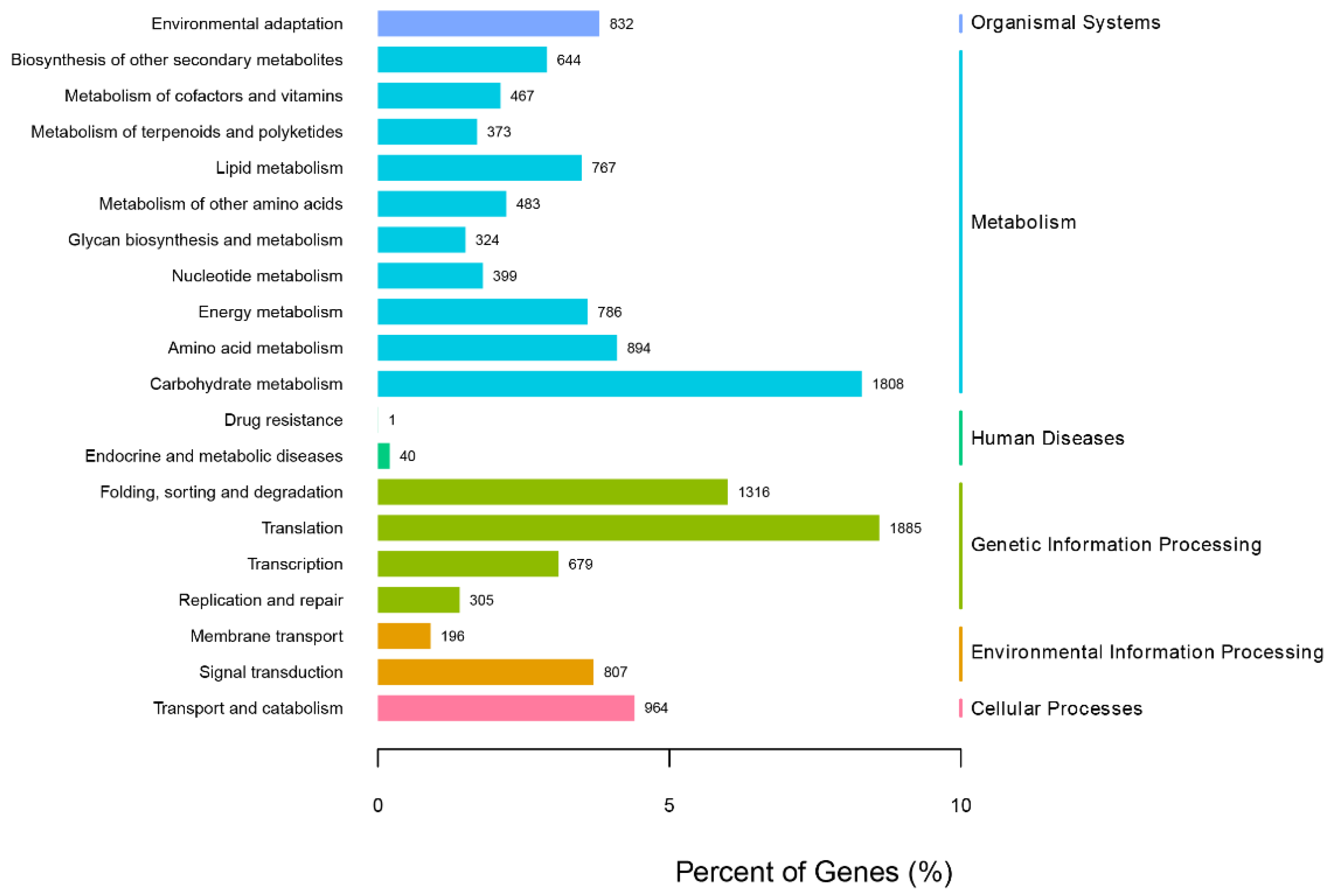
3.2. Functional Annotation of Unigenes
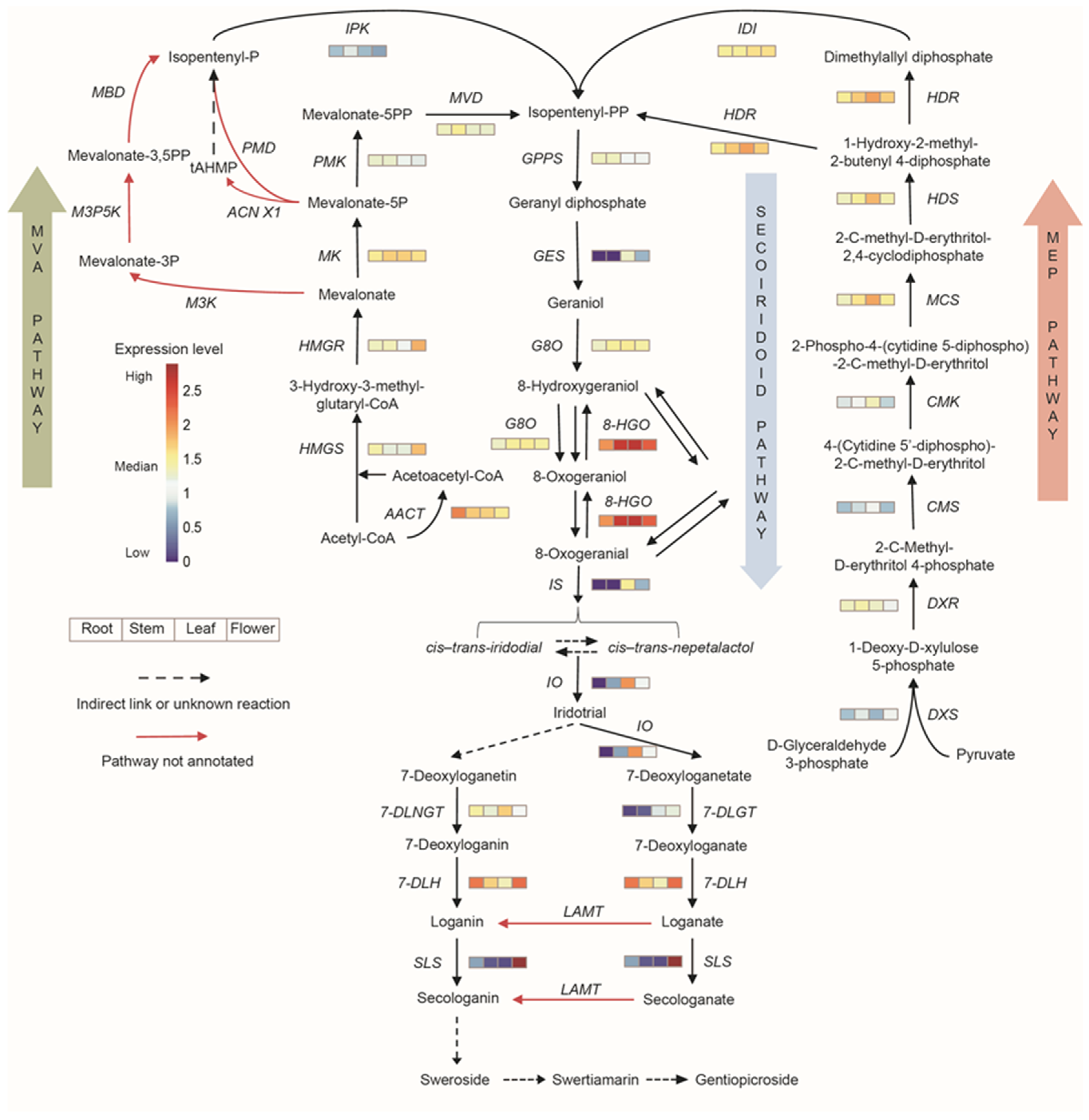
3.3. Correlation Analysis of Secondary Metabolism
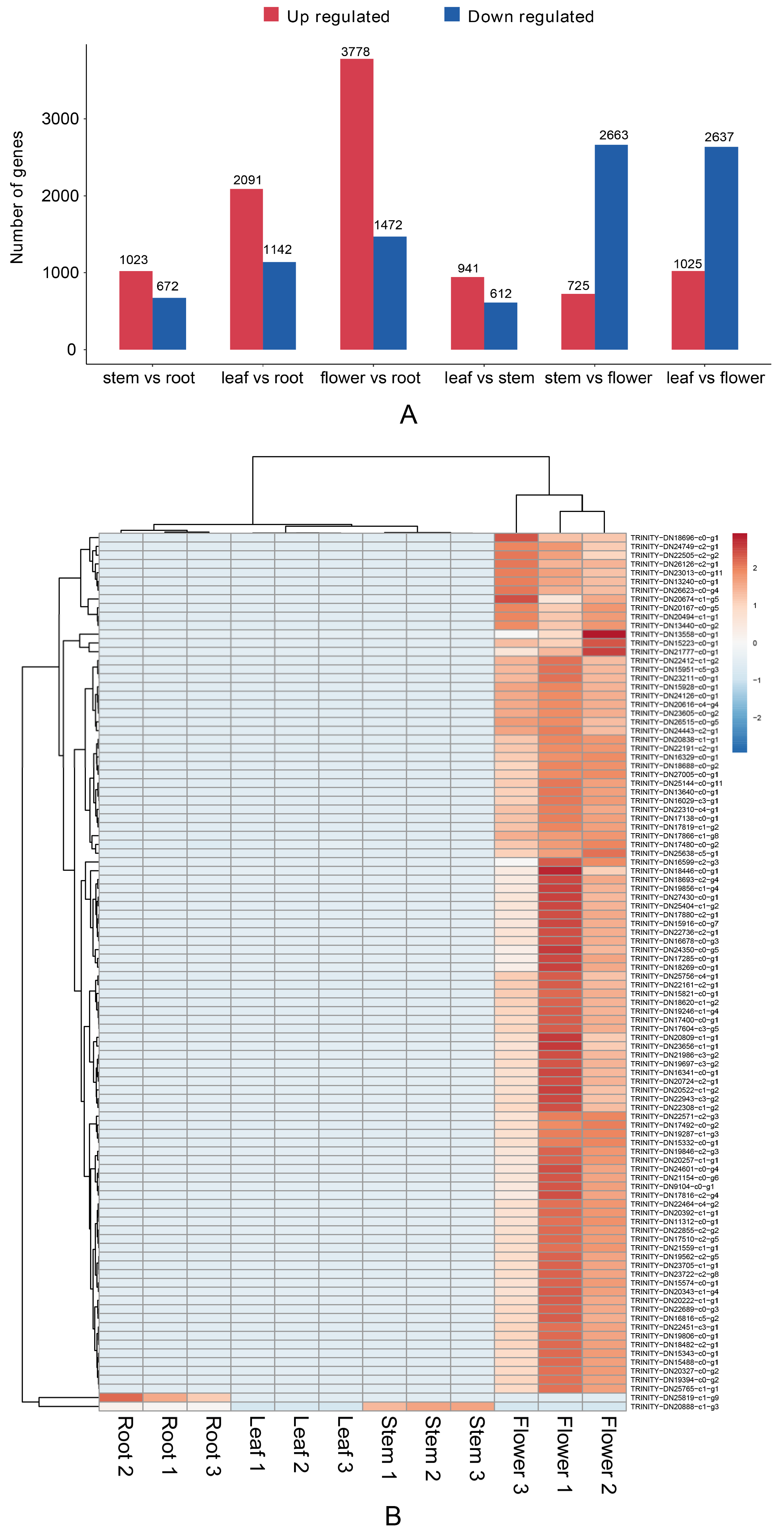
3.4. Differential Gene Analysis
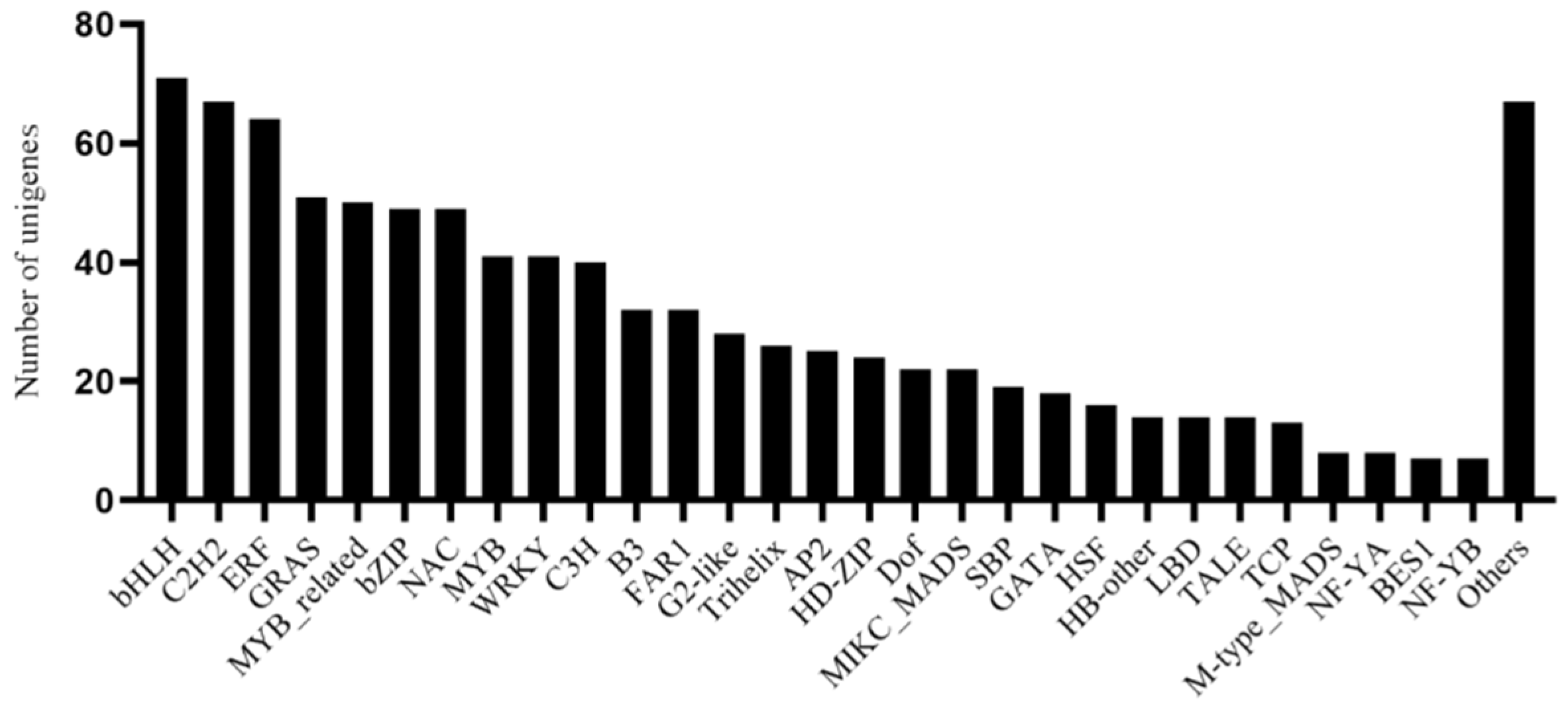
3.5. Analysis of Transcription Factors
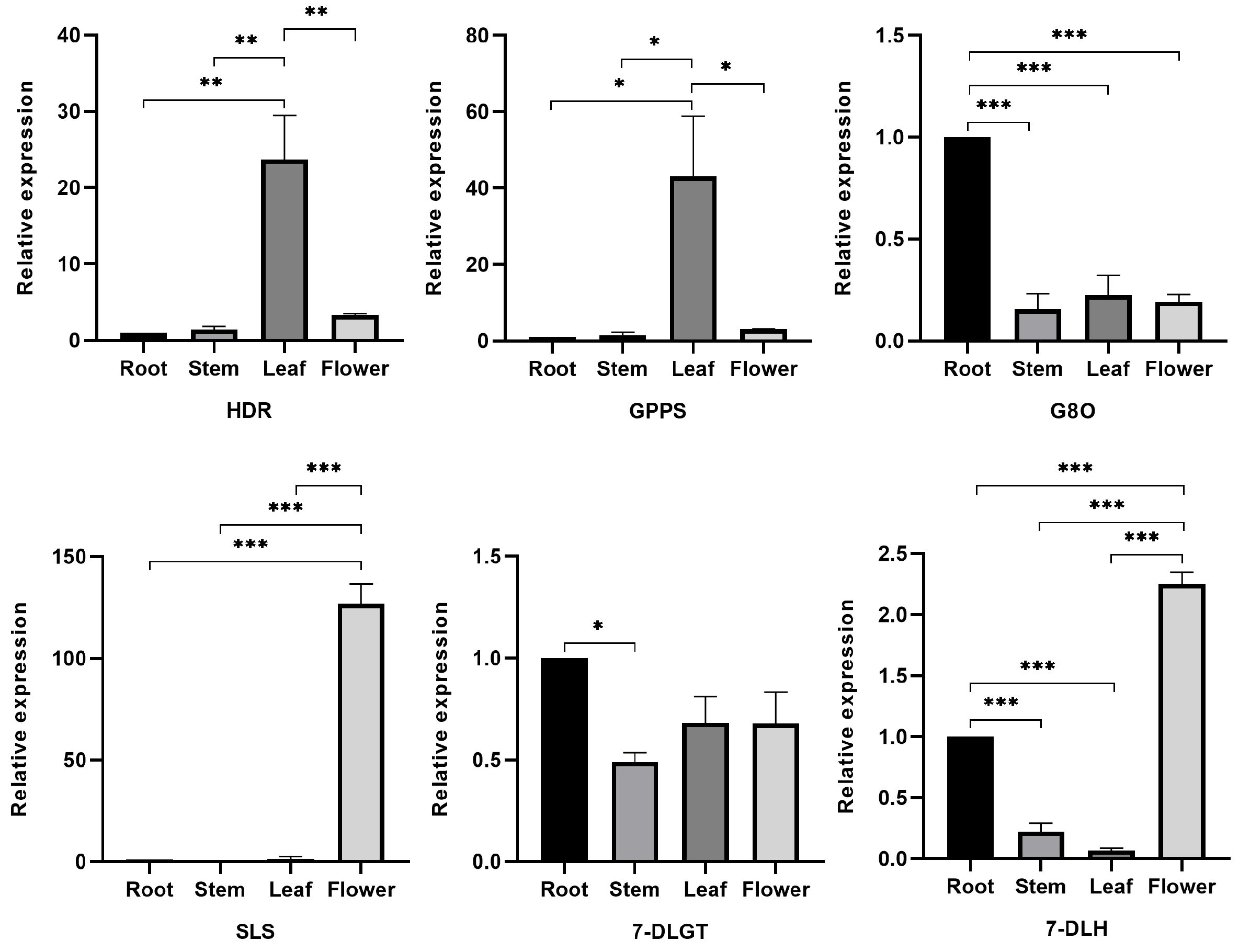
3.6. Validation of Key Enzyme Genes Using qRT-PCR
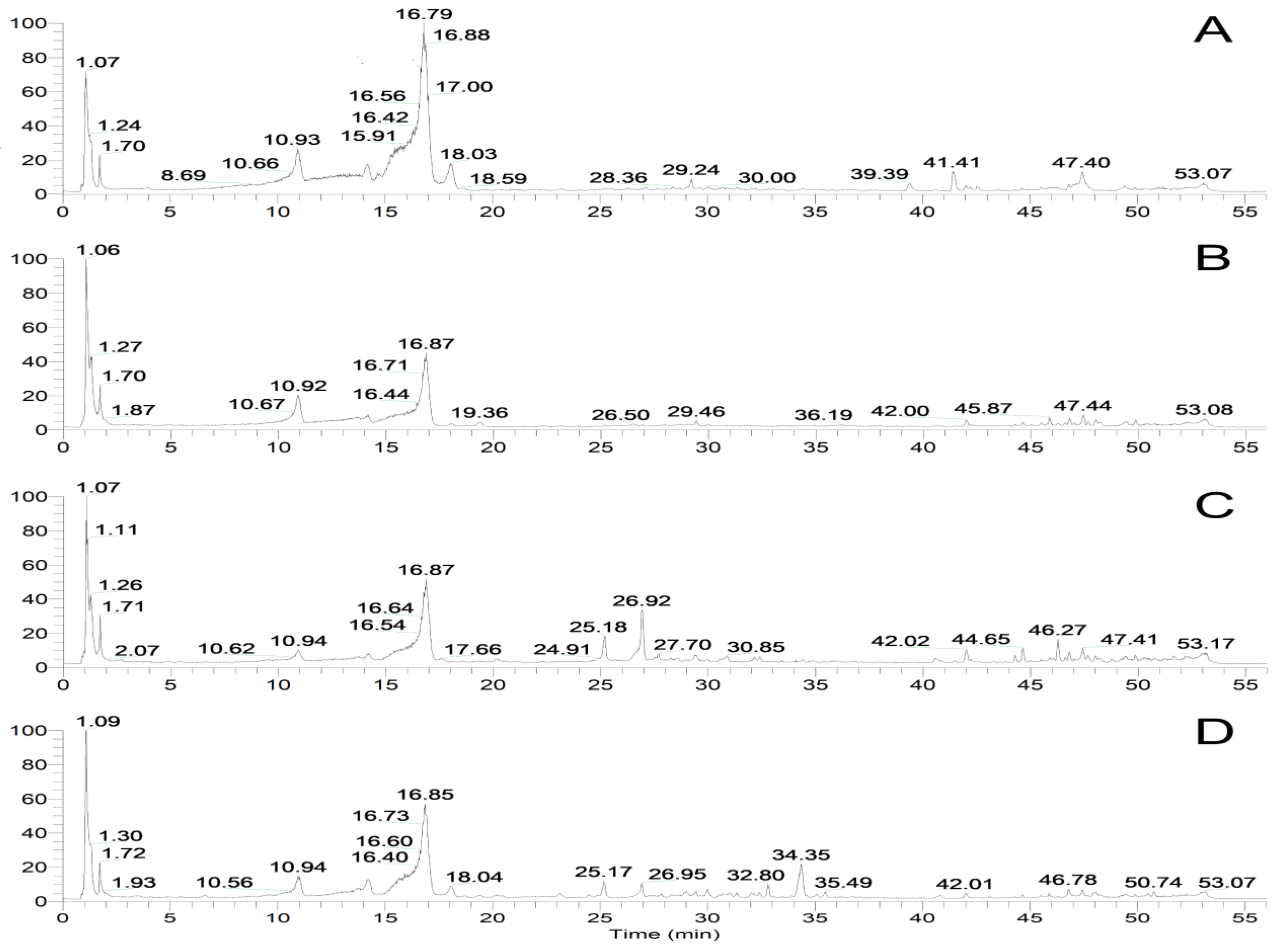
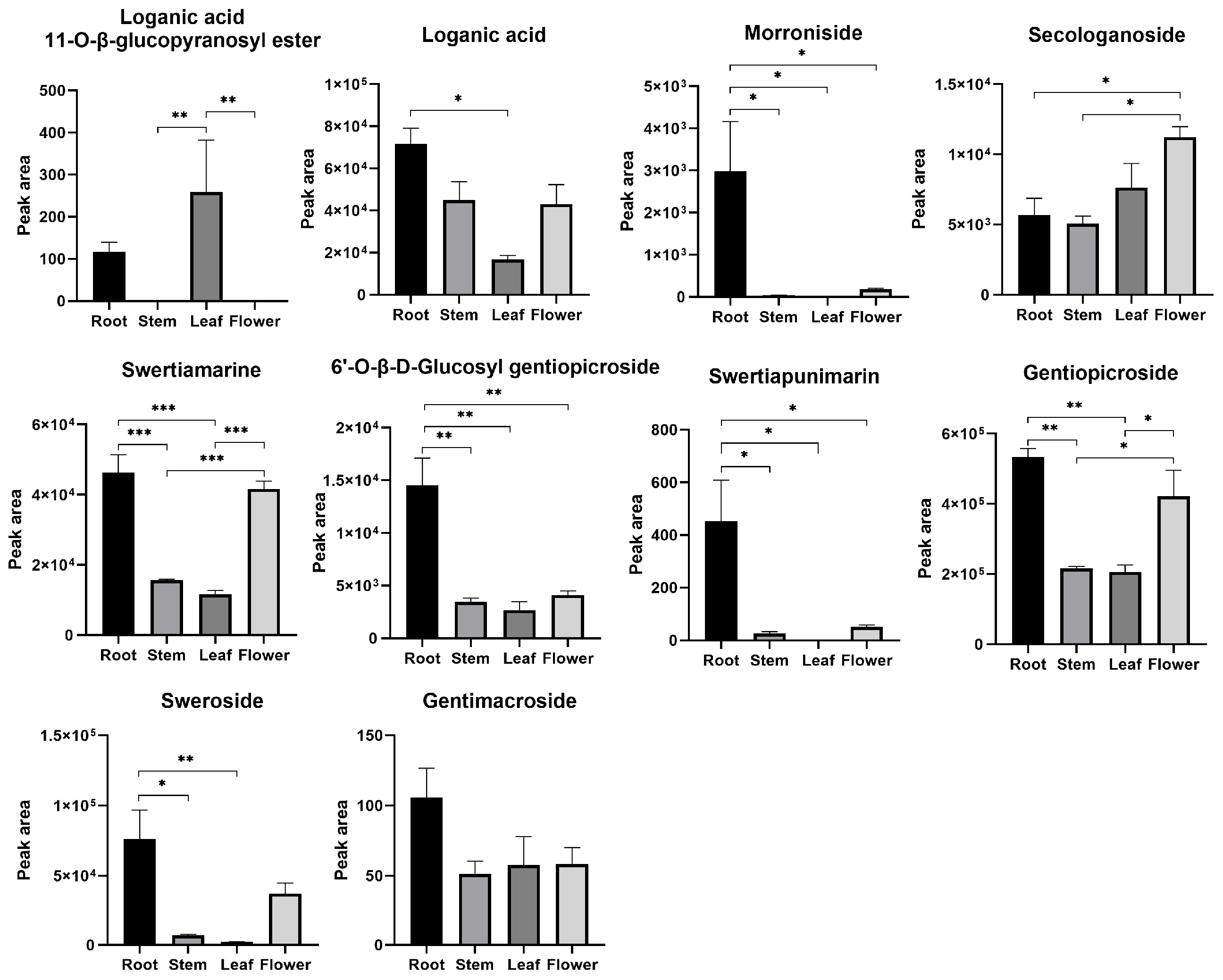
3.7. Metabolite Analysis of G. macrophylla by UPLC-Q-Exactive MS
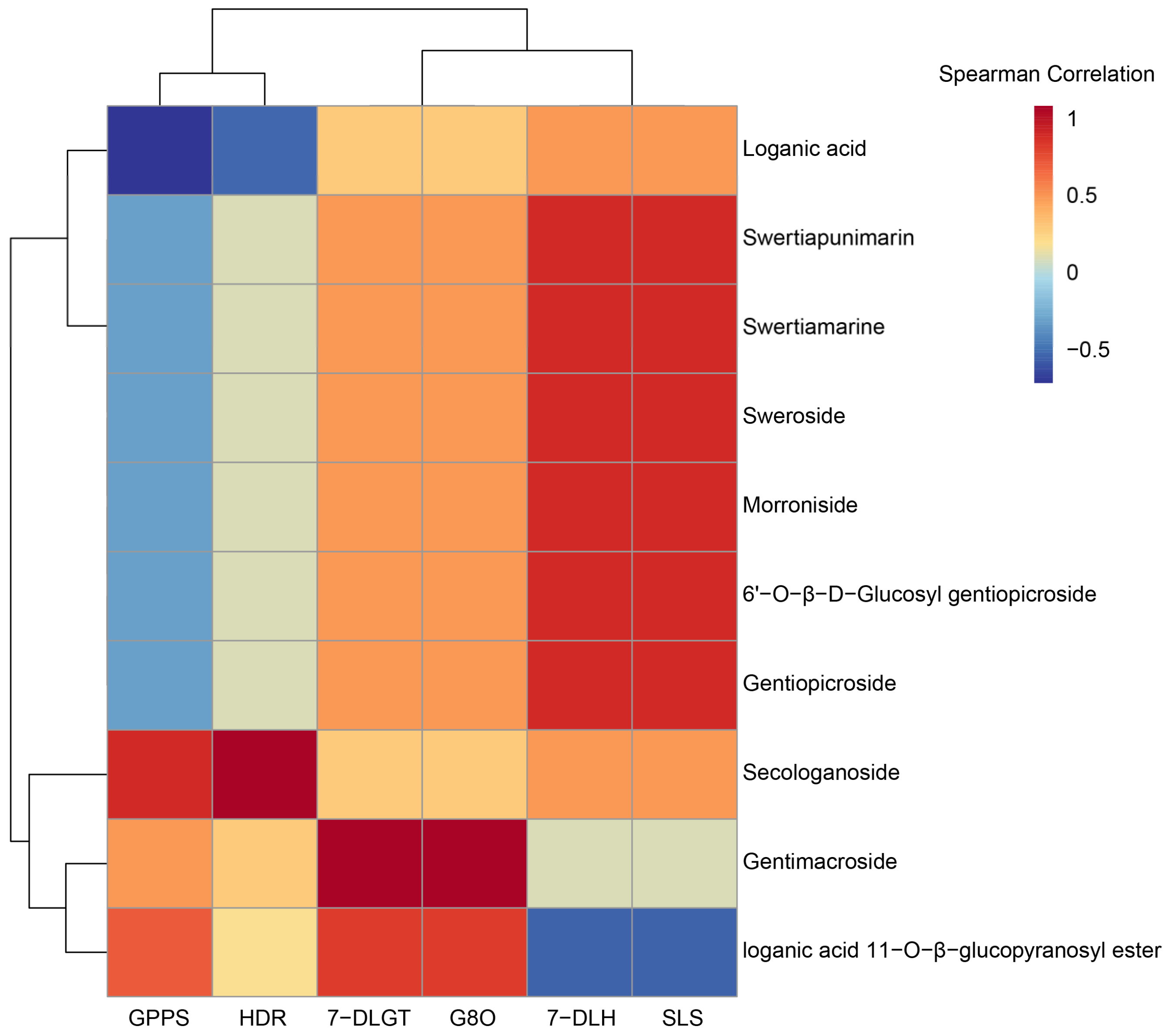
3.8. Correlation Analysis between the Expression of Key Enzyme Genes and Contents of Iridoids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, Y.Y.; Yang, Y.M.; Ma, X.H.; Zhang, X.B.; Zhu, S.D.; Jin, L. Ecology suitability study of Chinese materia medica Gentianae Macrophyllae Radix. China J. Chin. Mater. Med. 2016, 41, 3176–3180. [Google Scholar]
- Zhao, Z.L.; Dorje, G.; Wang, Z.T. Identification of medicinal plants used as Tibetan traditional medicine Jie-Ji. J. Ethnopharmacol. 2010, 132, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.B.; Liu, H.L.; Liu, X.Q.; Shang, J.N.; Zhao, J.R.; Yuan, C.S. Chemical constituents of Gentiana macrophylla Pall. Nat. Prod. Res. 2010, 24, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhao, Y.L.; Zhang, J.; Li, W.Y.; Wang, Y.Z. Phytochemistry and pharmacological activities of the genus Gentiana (Gentianaceae). Chem. Biodivers. 2016, 13, 107–150. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.H.; Zhang, X.J.; Yun, S.S.; Yu, J.H. Prediction of the ecological suitability of Gentiana macrophylla Pall. under scenarios of global climate change. Chin. J. Ecol. 2020, 39, 3766–3773. [Google Scholar]
- Tholl, D. Biosynthesis and biological functions of terpenoids in plants. Adv. Biochem. Eng./Biotechnol. 2015, 148, 63–106. [Google Scholar]
- Hua, W.; Zheng, P.; He, Y.; Cui, L.; Kong, W.; Wang, Z. An insight into the genes involved in secoiridoid biosynthesis in Gentiana macrophylla by RNA-seq. Mol. Biol. Rep. 2014, 41, 4817–4825. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Zou, L.; Sun, S.; Kuang, X.; Wei, J.; Wang, L.; Li, Y.; Sun, C. Hybrid sequencing of the Gynostemma pentaphyllum transcriptome provides new insights into gypenoside biosynthesis. BMC Genom. 2019, 20, 632–645. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Li, Z.; Li, Y.; Wang, S.; Li, L.; Liu, M.; Ye, J. Transcriptome analysis of terpenoid biosynthetic genes and simple sequence repeat marker screening in Eucommia ulmoides. Mol. Biol. Rep. 2020, 47, 1979–1990. [Google Scholar] [CrossRef]
- Hou, D.Y.; Shi, L.C.; Yang, M.M.; Li, J.; Zhou, S.; Zhang, H.X.; Xu, H.W. De novo transcriptomic analysis of leaf and fruit tissue of Cornus officinalis using Illumina platform. PLoS ONE 2018, 13, e0192610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.S.; Xing, Z.; Liu, H.; Hu, X.G.; Gao, Q.; Xu, J.; Jiao, S.Q.; Jia, K.H.; Jin, Y.Q.; Zhao, W.; et al. In-depth transcriptome characterization uncovers distinct gene family expansions for Cupressus gigantea important to this long-lived species’ adaptability to environmental cues. BMC Genom. 2019, 20, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafez, O.H.; Othman, E.M.; Fahim, J.R.; Desoukey, S.Y.; Pimentel-Elardo, S.M.; Nodwell, J.R.; Schirmeister, T.; Tawfike, A.; Abdelmohsen, U.R. Metabolomics analysis and biological investigation of three Malvaceae plants. Phytochem. Anal. 2020, 31, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Morreeuw, Z.P.; Escobedo-Fregoso, C.; Ríos-González, L.J.; Castillo-Quiroz, D.; Reyes, A.G. Transcriptome-based metabolic profiling of flavonoids in Agave lechuguilla waste biomass. Plant Sci. 2021, 305, 110748. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Dong, Y.; Lv, M.; Qian, L.; Sun, X.; Liu, L.; Cai, Y.; Fan, H. Combined Analysis of Volatile Terpenoid Metabolism and Transcriptome Reveals Transcription Factors Related to Terpene Synthase in Two Cultivars of Dendrobium officinale Flowers. Front. Genet. 2021, 12, 661296. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shi, Z.; Wang, Y.; Zhang, R.; Hu, H. Study on effects of sweating treatment and different drying methods on the quality of Gentianae macrophyllae Radix. Sci. Rep. 2021, 11, 9717. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boratyn, G.M.; Thierry-Mieg, J.; Thierry-Mieg, D.; Busby, B.; Madden, T.L. Magic-BLAST, an accurate RNA-seq aligner for long and short reads. BMC Bioinform. 2019, 20, 405. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2002, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Dong, J.H.; Bao, Y.T.; Chen, X.H.; Hai, Y.; Zeng, R. Rapid analysis on chemical constituents from roots of Gentiana crasicaulis by ultra-high performance liquid chromatography coupled with hybrid quadrupole-orbitrap mass spectrometry. Chin. Tradit. Herb. Drugs 2010, 47, 3175–3180. [Google Scholar]
- Zhang, Z.K.; Li, Y.Q.; Wang, Z.X.; Yang, X.X.; Xiao, Z.J.; Ma, C.H. Rapid analysis of 4 chemical compositions in Gentiana macrophylla by UPLC-LTQ-Orbitrap-MS. Cent. South Pharm. 2020, 18, 96–101. [Google Scholar]
- Wei, S.; Zhang, P.; Feng, X.; Kodama, H.; Yu, C.; Chen, G. Qualitative and quantitative determination of ten iridoids and secoiridoids in Gentiana straminea Maxim. by LC-UV-ESI-MS. J. Nat. Med. 2012, 66, 102–108. [Google Scholar] [CrossRef]
- Ahn, S.J.; Kim, H.J.; Lee, A.; Min, S.S.; In, S.; Kim, E. Determination of 12 herbal compounds for estimating the presence of Angelica Gigas Root, Cornus Fruit, Licorice Root, Pueraria Root, and Schisandra Fruit in foods by LC-MS/MS. Food Addit. Contam. Part A 2020, 37, 1437–1448. [Google Scholar] [CrossRef]
- Karioti, A.; Chatzopoulou, A.; Bilia, A.R.; Liakopoulos, G.; Stavrianakou, S.; Skaltsa, H. Novel secoiridoid glucosides in Olea europaea leaves suffering from boron deficiency. Biosci. Biotechnol. Biochem. 2006, 70, 1898–1903. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.L.; Sun, W.J.; Tu, G.Z.; Shi, Z.Y. Chemical Constituents of Gentiana Macrophylla Pall. From Shaanxi. Nat. Prod. Commun. 2006, 1, 527–530. [Google Scholar] [CrossRef]
- Chen, Q.L.; Shi, Z.Y.; Zhang, Y.H.; Zheng, J.B. Study on the chemical constituents in roots of Gentiana dahurica. J. Chin. Med. Mater. 2011, 8, 1214–1216. [Google Scholar]
- He, K.; Cao, T.W.; Wang, H.L.; Geng, C.A.; Zhang, X.M.; Chen, J.J. Chemical constituents of Swertia kouitchensis Franch. China J. Chin. Mater. Med. 2015, 40, 3811–3817. [Google Scholar]
- Duan, C.H.; Shi, B.J.; Wu, L.H.; Chou, G.X.; Wang, Z.T. Chemical Constituents of Gentiana waltonii. Chin. J. Nat. Med. 2007, 6, 417–420. [Google Scholar]
- Li, Y.W.; Li, R.J.; Yuan, L.Y.; Chang, Y.; Wang, X.L.; Chen, L.H. Chemical constituents from flower of Gentiana tibetica. Chin. Tradit. Herb. Drugs 2015, 46, 2052–2056. [Google Scholar]
- Chen, J.K.; Zeng, R. Application of metabolomics approach to study on chemical constituents in different parts of Gentiana crasicaulis based on UPLC-ESI-HRMSn. Chin. Tradit. Herb. Drugs 2018, 49, 2328–2335. [Google Scholar]
- Kouda, R.; Yakushiji, F. Recent Advances in Iridoid Chemistry: Biosynthesis and Chemical Synthesis. Chem. Asian J. 2020, 15, 3771–3783. [Google Scholar] [CrossRef]
- Kang, H.; Zhao, Z.L.; Ni, L.H.; Li, W.T.; Zhao, S.J.; Liu, T.H. Transcriptome analysis and validation of key genes involved in biosynthesis of iridoids in Gentiana lhassica. China J. Chin. Mater. Med. 2021, 46, 4704–4711. [Google Scholar]
- Ni, L.; Zhao, Z.; Wu, J.; Gaawe, D.; Mi, M. Analysis of transcriptomes to explore genes contributing to iridoid biosynthesis in Gentiana waltonii and Gentiana robusta (Gentianaceae). Acta Pharm. Sin. 2019, 54, 944–953. [Google Scholar]
- Hemmerlin, A. Post-translational events and modifications regulating plant enzymes involved in isoprenoid precursor biosynthesis. Plant Sci. 2013, 203–204, 41–54. [Google Scholar] [CrossRef]
- Zhou, T.; Bai, G.; Hu, Y.; Ruhsam, M.; Yang, Y.; Zhao, Y. De novo genome assembly of the medicinal plant Gentiana macrophylla provides insights into the genomic evolution and biosynthesis of iridoids. DNA Res. 2022, 5, dsac034. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.P.; Min, C.; Rong, C.; Xiao, Z.L.; Ming, H.H.; Liao, Z. The last gene involved in the MEP pathway of Artemisia annua: Cloning and characterization and functional identification. J. Med. Plants Res. 2011, 5, 223–230. [Google Scholar]
- Huang, J.Z.; Cheng, T.C.; Wen, P.J.; Hsieh, M.H.; Chen, F.C. Molecular characterization of the Oncidium orchid HDR gene encoding 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase, the last step of the methylerythritol phosphate pathway. Plant Cell Rep. 2009, 28, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.H.; Goodman, H.M. The Arabidopsis IspH homolog is involved in the plastid nonmevalonate pathway of isoprenoid biosynthesis. Plant Physiol. 2005, 138, 641–653. [Google Scholar] [CrossRef]
- National Pharmacopoeia Committee. Pharmacopoeia of Peoples Republic of China; Part 1; Chemical Industry Press: Beijing, China, 2020; p. 270. [Google Scholar]
- Morreeuw, Z.P.; Castillo-Quiroz, D.; Ríos-González, L.J.; Martínez-Rincón, R.; Estrada, N.; Melchor-Martínez, E.M.; Iqbal, H.M.N.; Parra-Saldívar, R.; Reyes, A.G. High Throughput Profiling of Flavonoid Abundance in Agave lechuguilla Residue-Valorizing under Explored Mexican Plant. Plants 2021, 10, 695. [Google Scholar] [CrossRef]
- Liu, L.; Zuo, Z.T.; Xu, F.R.; Wang, Y.Z. Study on Quality Response to Environmental Factors and Geographical Traceability of Wild Gentiana rigescens Franch. Front. Plant Sci. 2020, 11, 1128. [Google Scholar] [CrossRef]
- Leavitt, J.M.; Alper, H.S. Advances and current limitations in transcript-level control of gene expression. Curr. Opin. Biotechnol. 2015, 34, 98–104. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Pathway | Number of Unigenes | Proportion % | KEGG ID |
|---|---|---|---|---|
| 1 | Phenylpropanoid biosynthesis | 354 | 16.31 | K09753 |
| 2 | Flavonoid biosynthesis | 129 | 5.94 | K00660 |
| 3 | Ubiquinone and other terpenoid-quinone biosyntheses | 96 | 4.42 | K12501 |
| 4 | Terpenoid backbone biosynthesis | 92 | 4.24 | K05906 |
| 5 | Phenylalanine, tyrosine, and tryptophan biosynthesis | 78 | 3.59 | K00500 |
| 6 | Tropane, piperidine, and pyridine alkaloid biosynthesis | 65 | 3.00 | K08081 |
| 7 | Fatty acid biosynthesis | 64 | 2.95 | K00665 |
| 8 | Stilbenoid, diarylheptanoid, and gingerol biosynthesis | 63 | 2.90 | K13065 |
| 9 | Sesquiterpenoid and triterpenoid biosynthesis | 62 | 2.86 | K15803 |
| 10 | Cutin, suberine, and wax biosynthesis | 56 | 2.58 | K15403 |
| 11 | Isoquinoline alkaloid biosynthesis | 56 | 2.58 | K14454 |
| 12 | Biosynthesis of unsaturated fatty acids | 50 | 2.30 | K00507 |
| 13 | Diterpenoid biosynthesis | 48 | 2.21 | K04125 |
| 14 | Valine, leucine, and isoleucine biosynthesis | 33 | 1.52 | K00826 |
| 15 | Indole alkaloid biosynthesis | 26 | 1.20 | K21026 |
| 16 | Zeatin biosynthesis | 21 | 0.97 | K00279 |
| 17 | Monoterpenoid biosynthesis | 21 | 0.97 | K15095 |
| 18 | Monobactam biosynthesis | 18 | 0.83 | K12524 |
| 19 | Flavone and flavonol biosynthesis | 17 | 0.78 | K05280 |
| 20 | Benzoxazinoid biosynthesis | 16 | 0.74 | K13229 |
| 21 | Betalain biosynthesis | 15 | 0.69 | K15777 |
| 22 | Anthocyanin biosynthesis | 12 | 0.55 | K21383 |
| 23 | Phenazine biosynthesis | 7 | 0.32 | K01657 |
| No. | Ion Mode | tR/min | Excimer Ion Peaks | Molecular Formula | Adduct Ions | Ppm | Maior Fragment Ions (m/z) | Compounds | Parts | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Control | Measured (m/z) | Calculated (m/z) | ||||||||
| 1 | − | 1.10 | 341.1094 | 341.1089 | C12H22O11 | [M−H]− | 1.38 | 178.0560, 161.0448, 131.0339, 119.0335, 101.0229, 89.0228, 71.0122 | Sucrose [27] | R, S, L, F | |
| 2 | − | 1.68 | 191.0191 | 191.0197 | C6H8O7 | [M−H]− | −3.30 | 159.3513, 134.9712, 111.0074, 87.0072, 67.0173, 59.0122, 57.0330 | Citric Acid [27] | R, S, L, F | |
| 3 | − | 6.95 | 583.1870 | 583.1880 | C22H34O15 | [M+HCOO]− | −1.66 | 375.1304, 313.1309, 213.0761, 169.0865, 113.0229, 59.0122 | Loganic acid 11-O-β-glucopyranosyl ester [4] | R, L | |
| 4 | − | 10.98 | 10.90 | 375.1299 | 375.1297 | C16H24O10 | [M−H]− | 0.61 | 213.0764, 169.0861, 151.0754, 133.0647, 113.0231, 95.0487, 69.0329 | Loganic acid [28] | R, S, L, F |
| 5 | + | 11.69 | 429.1366 | 429.1367 | C17H26O11 | [M−Na]− | −0.23 | 267.0845, 235.0575, 203.0521, 185.0418, 110.6607, 79.2134 | Morroniside [29] | R, S, F | |
| 6 | − | 13.78 | 389.1104 | 389.1089 | C16H22O11 | [M−H]− | 3.78 | 319.0093, 2199.2400, 183.0659, 165.0549, 121.0645, 69.0329, 59.0122 | Secologanoside [30] | R, S, L, F | |
| 7 | − | 14.21 | 14.16 | 419.1201 | 419.1195 | C16H22O10 | [M+HCOO]− | 1.43 | 302.6173, 179.0552, 149.0597, 141.0181, 119.0336, 113.0230, 89.0228, 59.0122 | Swertiamarine [31] | R, S, L, F |
| 8 | − | 14.71 | 14.60 | 563.1631 | 563.1618 | C22H30O14 | [M+HCOO]− | 2.38 | 221.0665, 193.0498, 179.0552, 161.0445, 131.0337, 101.0229, 89.0228 | 6’-O-β-D-Glucosyl gentiopicroside [31] | R, S, L, F |
| 9 | + | 15.66 | 521.1862 | 521.1865 | C22H32O14 | [M+H]+ | −0.58 | 251.4391, 197.0808, 179.0703, 151.0751, 127.0389, 111.0801 | Swertiapunimarin [32] | R, S, F | |
| 10 | − | 16.79 | 16.83 | 401.1093 | 401.1089 | C16H20O9 | [M+HCOO]− | 0.92 | 324.6015, 219.0556, 149.0597, 121.0648, 113.0231, 93.0333, 89.0228 | Gentiopicroside [31] | R, S, L, F |
| 11 | + | 16.81 | 177.0545 | 177.0546 | C10H8O3 | [M+H]+ | −0.68 | 147.0440, 131.0491, 121.0647, 119.0497, 103.0543, 91.0492, 79.0544 | Erythrocentaurine [33] | R, S, L, F | |
| 12 | + | 16.81 | 195.0649 | 195.0652 | C10H10O4 | [M+H]+ | −1.49 | 177.0549, 149.0598, 131.0490, 121.0648, 103.0542, 91.0543, 79.0543 | Ferulic acid [34] | R, S, L, F | |
| 13 | − | 18.10 | 18.23 | 403.1254 | 403.1246 | C16H22O9 | [M+HCOO]− | 2.03 | 312.8494, 205.4160, 151.0749, 125.0229, 89.0229, 81.0329 | Sweroside [31] | R, S, L, F |
| 14 | − | 20.10 | 593.1524 | 593.1512 | C27H30O15 | [M−H]− | 2.04 | 557.1314, 473.1095, 431.1017, 341.0662, 311.0567, 282.0520 | Saponarin [35] | R, S, L, F | |
| 15 | − | 26.94 | 431.0991 | 431.0984 | C21H20O10 | [M−H]− | 1.69 | 341.0669, 311.0566, 283.0620, 269.0458, 239.0713, 163.0392, 117.0332 | Isovitexin [35] | R, S, L, F | |
| 16 | + | 29.26 | 235.0961 | 235.0965 | C13H14O4 | [M+H]+ | −1.66 | 217.0858, 189.0907, 174.0675, 159.0804, 145.0647, 129.0698, 91.0542 | 2-methoxyanofinic acid [34] | R, S, L, F | |
| 17 | − | 29.27 | 603.1943 | 603.1931 | C25H34O14 | [M+HCOO]− | 2.06 | 323.0986, 263.0774, 189.0912, 161.0961, 119.0336, 101.0229, 89.0228 | Macrophylloside D [31] | R, S, L, F | |
| 18 | − | 33.09 | 301.0357 | 301.0354 | C15H10O7 | [M−H]− | 1.06 | 193.0136, 151.0024, 149.0233, 107.0124, 83.0121 | Quercetin [30] | R, S, L, F | |
| 19 | − | 35.35 | 285.0407 | 285.0405 | C15H10O6 | [M−H]− | 0.84 | 257.0463, 192.0056, 159.0079, 151.0025, 108.0200, 83.0122 | Kaempferol [30] | R, S, L, F | |
| 20 | − | 36.72 | 531.1522 | 531.1508 | C26H28O12 | [M−H]− | 2.64 | 315.0727, 297.0625, 189.0910, 153.0182, 109.0280 | Gentimacroside[3] | R, S, L, F | |
| 21 | − | 48.00 | 471.3488 | 471.3480 | C30H48O4 | [M−H]− | 1.74 | 471.3486, 439.8552, 218.8751, 101.9868, 79.8463 | Corosolic acid [36] | R, S, L, F | |
| 22 | − | 51.76 | 455.3539 | 455.3531 | C30H48O3 | [M−H]− | 1.82 | 455.3540, 229.2049, 177.5234, 151.2601, 141.7751 | Oleanic acid [36] | R, S, L, F | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kou, Y.; Yi, X.; Li, Z.; Ai, Y.; Ma, S.; Chen, Q. A Comparative Transcriptomic with UPLC-Q-Exactive MS Reveals Differences in Gene Expression and Components of Iridoid Biosynthesis in Various Parts of Gentiana macrophylla. Genes 2022, 13, 2372. https://doi.org/10.3390/genes13122372
Kou Y, Yi X, Li Z, Ai Y, Ma S, Chen Q. A Comparative Transcriptomic with UPLC-Q-Exactive MS Reveals Differences in Gene Expression and Components of Iridoid Biosynthesis in Various Parts of Gentiana macrophylla. Genes. 2022; 13(12):2372. https://doi.org/10.3390/genes13122372
Chicago/Turabian StyleKou, Yuhang, Xiaoying Yi, Zhuo Li, Yun Ai, Siting Ma, and Qianliang Chen. 2022. "A Comparative Transcriptomic with UPLC-Q-Exactive MS Reveals Differences in Gene Expression and Components of Iridoid Biosynthesis in Various Parts of Gentiana macrophylla" Genes 13, no. 12: 2372. https://doi.org/10.3390/genes13122372