
The Complete Chloroplast Genome Sequence of Machilus chuanchienensis (Lauraceae): Genome Structure and Phylogenetic Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials and DNA Extraction
2.2. DNA Sequencing, Genome Assembly and Annotations
2.3. Simple Sequence Repeats (SSRs) and Repeat Sequences Analysis
2.4. Putative RNA Editing Site and Codon Usage
2.5. Genomic Comparison with Other Species in Machilus
2.6. Phylogenetic Analysis
3. Results
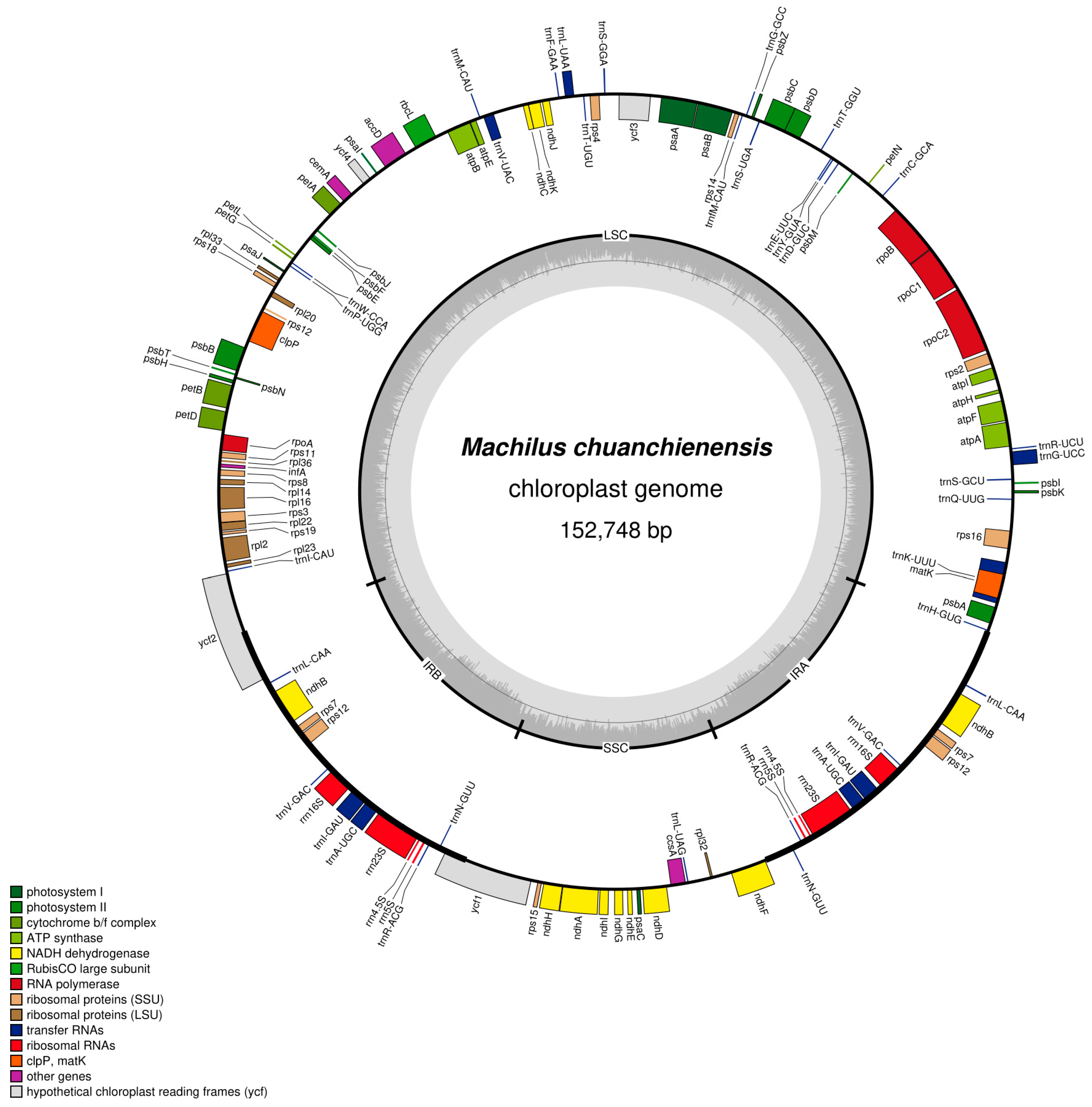
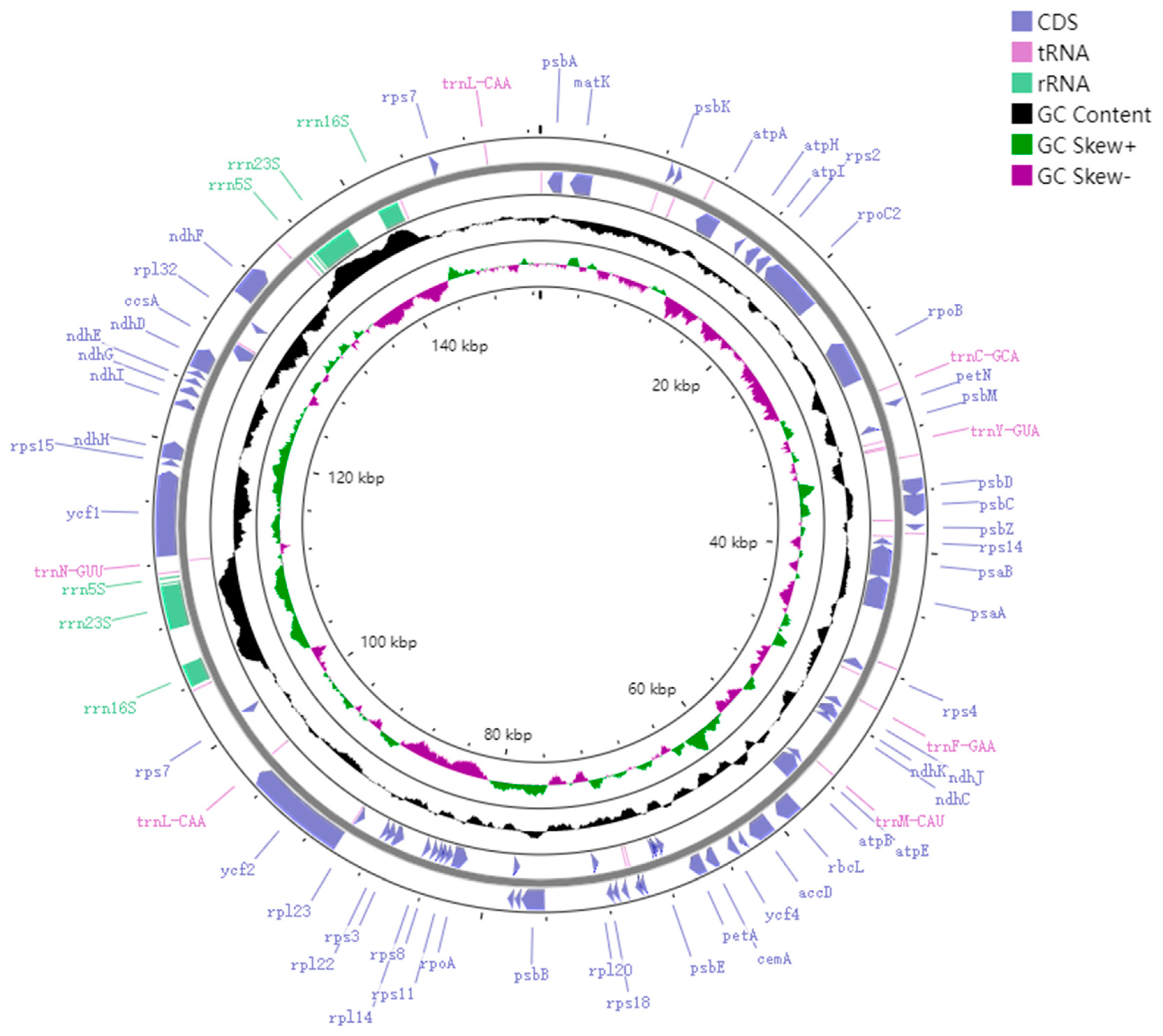
3.1. Structure and Characteristics of the M. chuanchienensis Chloroplast Genome
3.2. Analysis of SSRs and Long Repeats
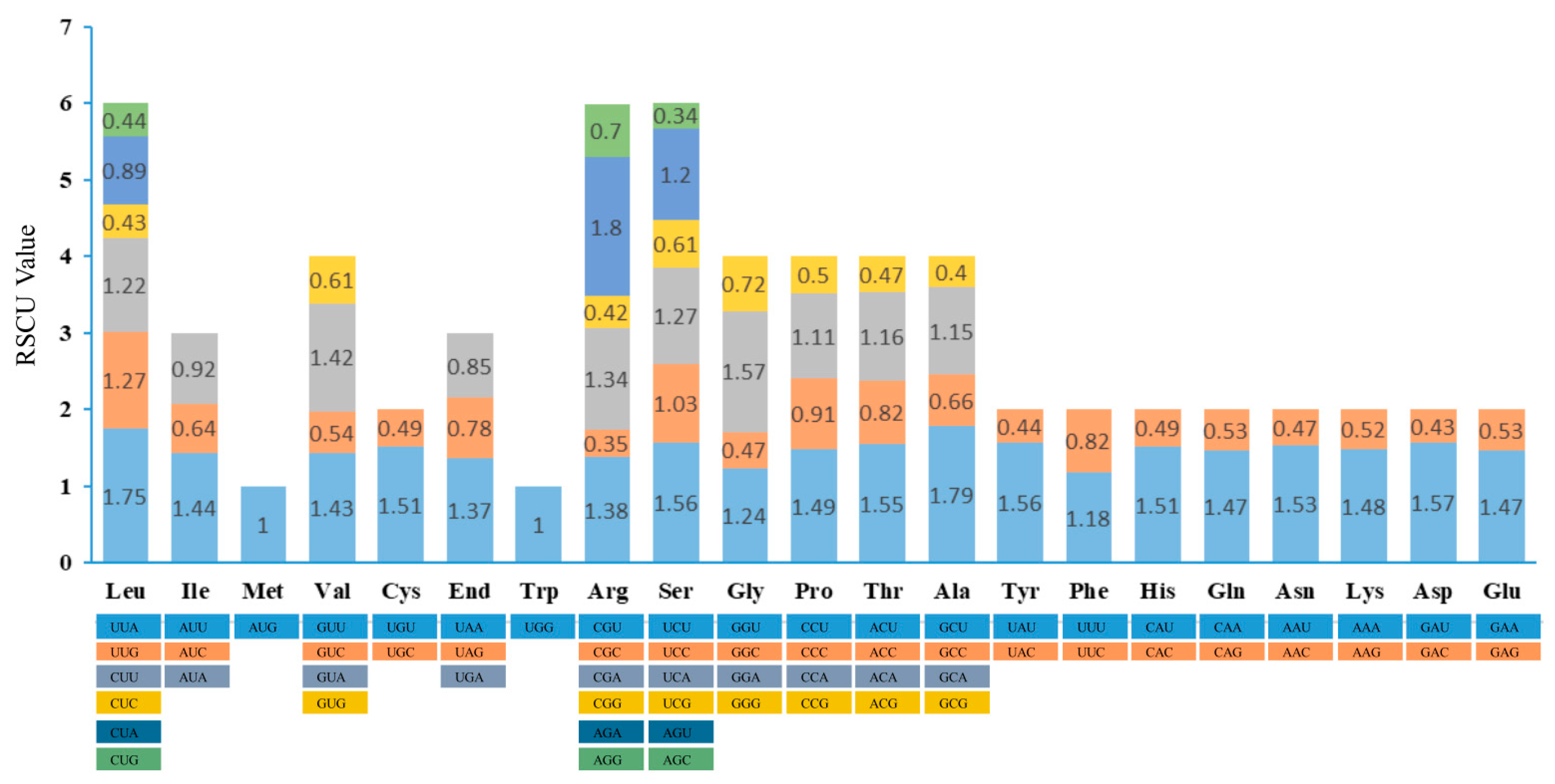
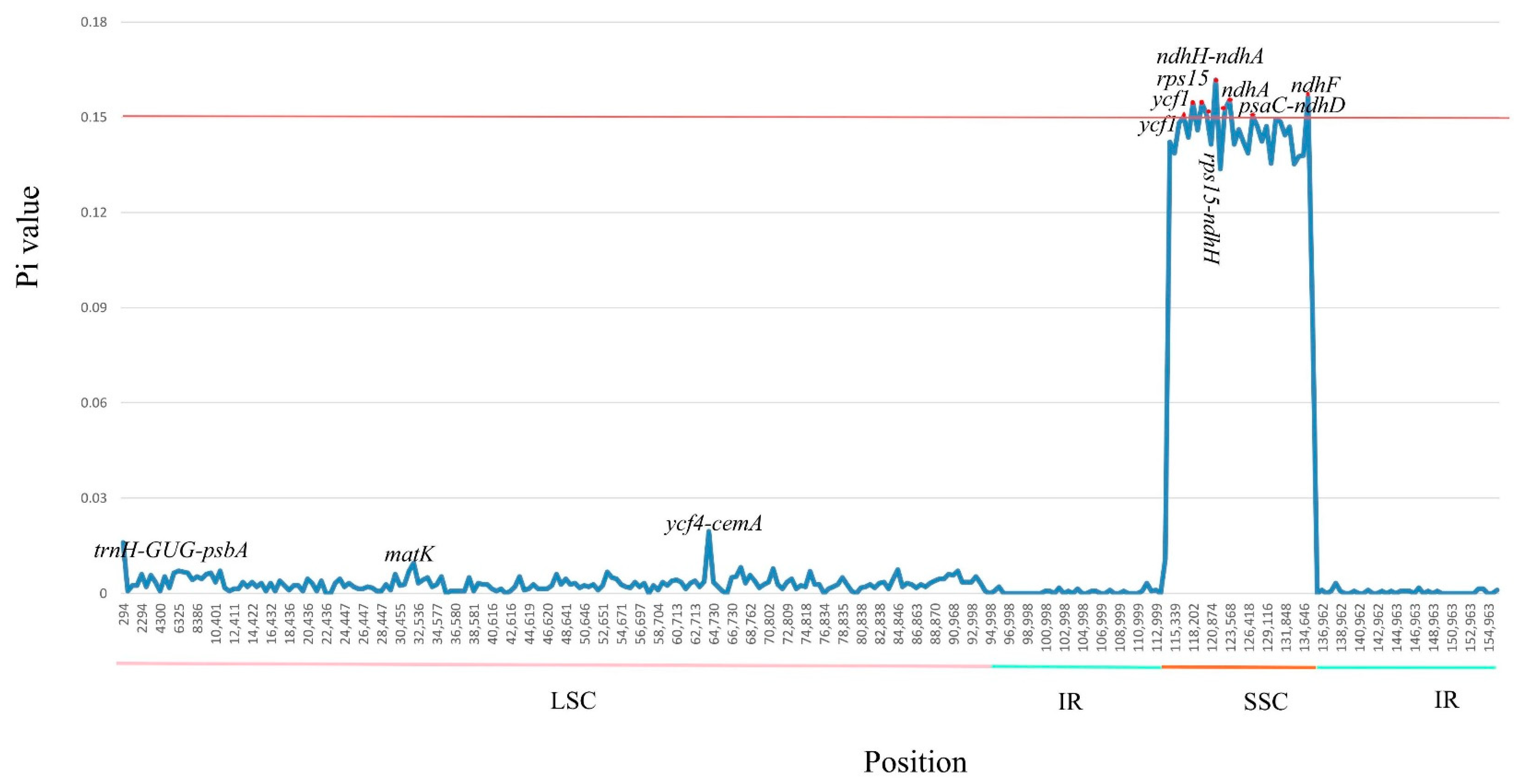
3.3. Codon Usage and Putative RNA Editing Site within M. chuanchienensis
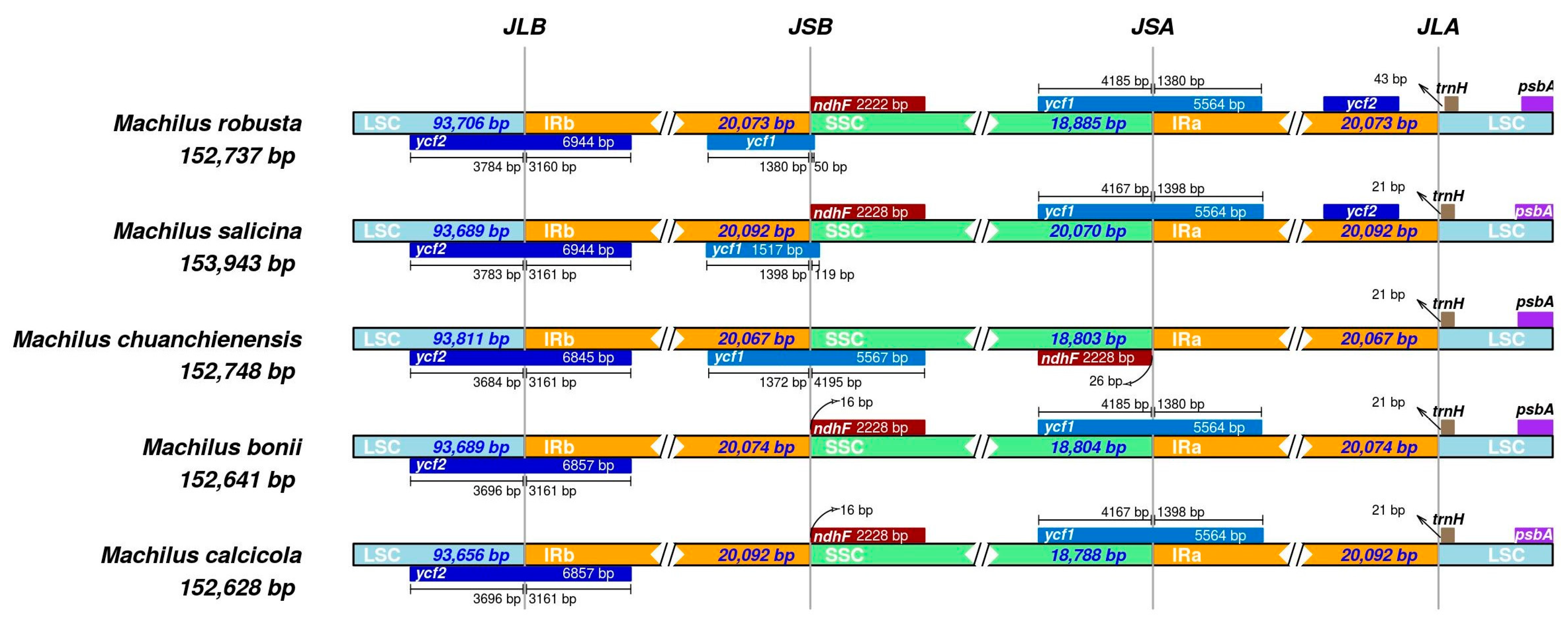
3.4. Genomic Comparison with Other Species in Machilus
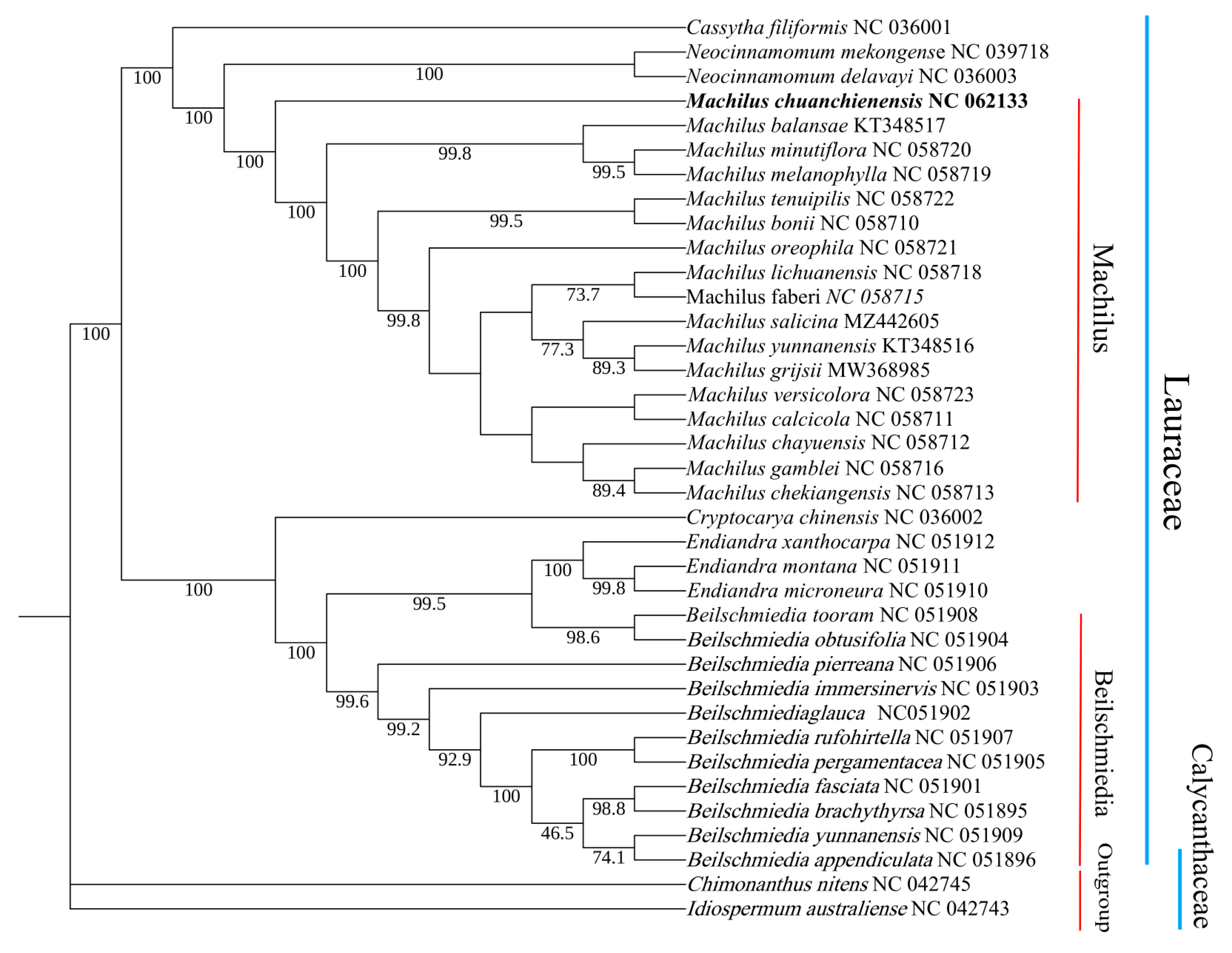
3.5. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.-C.; Chan, P.; Hsu, F.-L.; Chen, Y.-J.; Hsieh, M.-H.; Lo, M.-Y.; Lin, J.-Y. The In vitro inhibitory effects of crude extracts of traditional Chinese herbs on 3-Hydroxy-3-Methylglutaryl-Coenzyme a reductase on Vero cells. Am. J. Chin. Med. 2002, 30, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.-H.; Zhang, D.; Wang, G.; Yu, B.; Zhao, S.-P.; Wang, J.-W.; Yao, L.; Cao, W.-G. Comparative analyses of flavonoids compositions and antioxidant activities of Hawk tea from six botanical origins. Ind. Crops Prod. 2015, 80, 123–130. [Google Scholar] [CrossRef]
- Jia, X.; Dong, L.; Yang, Y.; Yuan, S.; Zhang, Z.; Yuan, M. Preliminary structural characterization and antioxidant activities of polysaccharides extracted from Hawk tea (Litsea coreana var. lanuginosa). Carbohydr. Polym. 2013, 95, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Zhang, Q.; Li, J.; Sun, Y.X.; Wang, L.Y.; Cheng, W.M.; Hu, X.Y. Antidiabetic effects of total flavonoids from Litsea Coreana leve on fat-fed, streptozotocin-induced type 2 diabetic rats. Am. J. Chin. Med. 2010, 38, 713–725. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Chen, Q.; Luo, L.; Ma, M.; Xiao, B.; Zeng, L. Camellia sinensis and Litsea coreana Ameliorate Intestinal Inflammation and Modulate Gut Microbiota in Dextran Sulfate Sodium-Induced Colitis Mice. Mol. Nutr. Food Res. 2020, 64, e1900943. [Google Scholar] [CrossRef]
- Wei, C.; He, P.; He, L.; Ye, X.; Cheng, J.; Wang, Y.; Li, W.; Liu, Y. Structure characterization and biological activities of a pectic polysaccharide from cupule of Castanea henryi. Int. J. Biol. Macromol. 2018, 109, 65–75. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, D.; Guo, J.-J.; Tao, W.; Gong, R.-X.; Yao, L.; Zhang, X.-L.; Cao, W.-G. Active Components, Antioxidant, Inhibition on Metabolic Syndrome Related Enzymes, and Monthly Variations in Mature Leaf Hawk Tea. Molecules 2019, 24, 657. [Google Scholar] [CrossRef] [Green Version]
- Chanderbali, A.S.; Werff, H.; Renner, S. Phylogeny and historical biogeography of Lauraceae: Evidence from the chloroplast and nuclear genomes. Ann. Mo. Bot. Gard. 2001, 88, 104–134. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Madriñán, S.; Li, J. Phylogeny and biogeography of Caryodaphnopsis (Lauraceae) inferred from low-copy nuclear gene and ITS sequences. Taxon 2016, 65, 433–443. [Google Scholar] [CrossRef]
- Song, Y.; Yao, X.; Tan, Y.; Gan, Y.; Corlett, R. Complete chloroplast genome sequence of the avocado: Gene organization, comparative analysis, and phylogenetic relationships with other Lauraceae. Can. J. For. Res. 2016, 46, 1293–1301. [Google Scholar] [CrossRef]
- Jia, X.J.; Ding, C.B.; Yuan, S.; Zhang, Z.W.; Chen, Y.E.; Du, L.; Yuan, M. Extraction, purification and characterization of polysaccharides from Hawk tea. Carbohydr. Polym. 2014, 99, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Na, W.; Liu, Z.; Chen, X.; Su, X. A novel turn-on fluorescent strategy for sensing ascorbic acid using graphene quantum dots as fluorescent probe. Biosens. Bioelectron. 2017, 92, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Hayashida, N.; Sugiura, M. Nicotiana chloroplast genes for components of the photosynthetic apparatus. Photosynth. Res. 1988, 18, 7–31. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Baurens, F.-C.; Cardi, C.; Aury, J.-M.; D’Hont, A. The Complete Chloroplast Genome of Banana (Musa acuminata, Zingiberales): Insight into Plastid Monocotyledon Evolution. PLoS ONE 2013, 8, e67350. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.C.; Zhang, Y.Z.; Geng, H.M.; Chen, S.L. The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 2016, 4, e2540. [Google Scholar]
- Gray, M.; Sankoff, D.; Cedergren, R.J. On the evolutionary descent of organisms and organelles: A global phylogeny based on a highly conserved structural core in small subunit ribosomal RNA. Nucleic Acids Res. 1984, 12, 5837–5852. [Google Scholar] [CrossRef]
- Sloan, D.B.; Triant, D.A.; Forrester, N.J.; Bergner, L.M.; Wu, M.; Taylor, D.R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenetics Evol. 2013, 72, 82–89. [Google Scholar] [CrossRef]
- Raman, G.; Park, S. The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis. Agronomy 2020, 10, 1405. [Google Scholar] [CrossRef]
- Song, Y.; Dong, W.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-F.; Ma, H.; Ci, X.-Q.; Li, L.; Song, Y.; Liu, B.; Li, H.-W.; Wang, S.-L.; Qu, X.-J.; Hu, J.-L.; et al. Can plastid genome sequencing be used for species identification in Lauraceae? Bot. J. Linn. Soc. 2021, 197, 1–14. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus. 1990, 12, 13–15. [Google Scholar]
- Wang, Y.; Wang, S.; Liu, Y.; Yuan, Q.; Sun, J.; Guo, L. Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genom. 2021, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kurt, S. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2011, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sylvester, S.P.; Li, M.; Zhang, C.; Li, X.; Duan, Y.; Wang, X. The Complete Plastid Genome of Magnolia zenii and Genetic Comparison to Magnoliaceae species. Molecules 2019, 24, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-Y.; Yu, Y.; Deng, Y.-Q.; Li, J.; Huang, Z.-X.; Zhou, S.-D. The Chloroplast Genome of Lilium henrici: Genome Structure and Comparative Analysis. Molecules 2018, 23, 1276. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; He, P.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37 (Suppl. S2), W253–W259. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wen, D.; Yu, Y.; Meudt, H.M.; Nakhleh, L. Bayesian inference of phylogenetic networks from bi-allelic genetic markers. PLOS Comput. Biol. 2018, 14, e1005932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, C.; Miao, H.; Xiong, S. Insights from the Complete Chloroplast Genome into the Evolution of Sesamum indicum L. PLoS ONE 2013, 8, e80508. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.F.; Zheng, Y.G.; Liu, X.L.; Dai, X.Y.; Li, S.X.; Xu, H. The complete chloroplast genome sequence of Machilus robusta W. W. Smith (Lauraceae) from Jiangxi Province, China. Mitochondrial DNA Part B 2021, 6, 1890–1892. [Google Scholar] [CrossRef] [PubMed]
- Xiaoxuan, L.; Shi, W.B.; Song, W.C.; Han, W.Q.; Yang, G.W.; Wang, S. Complete chloroplast genome sequence and annotation of Machilus salicina Hance, 1885 (lauraceae). Mitochondrial DNA Part B 2022, 7, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-B.; Yang, S.-X.; Li, H.T.; Yang, J.; Li, D.-Z. Comparative Chloroplast Genomes of Camellia Species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef] [Green Version]
- Raman, G.; Park, V.; Kwak, M.; Lee, B.; Park, S. Characterization of the complete chloroplast genome of Arabis stellari and comparisons with related species. PLoS ONE 2017, 12, e0183197. [Google Scholar] [CrossRef] [Green Version]
- Raman, G.; Choi, K.S.; Park, S. Phylogenetic Relationships of the Fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo Island Based on Chloroplast Genome Sequencing. Genes 2016, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Li, X.; Chen, X.; Huang, X.; Jin, S. The Complete Chloroplast Genome of Carya cathayensis and Phylogenetic Analysis. Genes 2022, 13, 369. [Google Scholar] [CrossRef]
- Yu, A.Y.H.; Houry, W.A. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007, 581, 3749–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, E.; Takahashi, Y.; Lemieux, C.; Turmel, M.; Rochaix, J. The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO J. 1997, 16, 6095–6104. [Google Scholar] [CrossRef] [PubMed]
- Naver, H.; Boudreau, E.; Rochaix, J.-D. Functional Studies of Ycf3: Its Role in Assembly of Photosystem I and Interactions with Some of Its Subunits. Plant Cell 2001, 13, 2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, I.; Matthews, P.J.; Biggs, P.J.; Naeem, M.; McLenachan, P.A.; Lockhart, P.J. Data from: Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Cavender-Bares, J.; González-Rodríguez, A.; Eaton, D.A.R.; Hipp, A.A.L.; Beulke, A.; Manos, P.S. Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Mol. Ecol. 2015, 24, 3668–3687. [Google Scholar] [CrossRef]
- Biju, V.C.; Shidhi, P.R.; Vijayan, S.; Rajan, V.S.; Sasi, A.; Janardhanan, A.; Nair, A.S. The Complete Chloroplast Genome of Trichopus zeylanicus, and Phylogenetic Analysis with Dioscoreales. Plant Genome 2019, 12, 190032. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.-Y.; Wu, H.; Wang, Y.-L.; Gao, L.-M.; Zhang, S.-Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.-H.; Kim, S.-C. Comparative Analysis of the Complete Chloroplast Genome Sequences of Three Closely Related East-Asian Wild Roses (Rosa sect. Synstylae; Rosaceae). Genes 2019, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, S.I.; Mehmood, F.; Ali, Z.; Malik, M.S.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 2019, 19, 100199. [Google Scholar] [CrossRef]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of the Medicinal Plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, H.-Y.; Zhang, Y.-H.; Yan, H.-J.; Qiu, X.-Q.; Wang, Q.-G.; Li, S.-B.; Zhang, S.-D. The Complete Chloroplast Genome of a Key Ancestor of Modern Roses, Rosa chinensis var. spontanea, and a Comparison with Congeneric Species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef] [Green Version]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef] [Green Version]
- Drescher, A.; Ruf, S.; Calsa, T., Jr.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef]
- Zhu, B.; Qian, F.; Hou, Y.; Yang, W.; Cai, M.; Wu, X. Complete chloroplast genome features and phylogenetic analysis of Eruca sativa (Brassicaceae). PLoS ONE 2021, 16, e0248556. [Google Scholar] [CrossRef]
- Li, B.; Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Gan, C.C.; Wang, H.C. Characteristics of the Dendrobium thyrsiflorum and its phylogenetic relationship analysis. Biotechnol. Bull. 2021, 37, 38–47. [Google Scholar]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef]
- Zhang, J.-B.; Li, R.-Q.; Xiang, X.-G.; Manchester, S.R.; Lin, L.; Wang, W.; Wen, J.; Chen, Z.-D. Integrated Fossil and Molecular Data Reveal the Biogeographic Diversification of the Eastern Asian-Eastern North American Disjunct Hickory Genus (Carya Nutt.). PLoS ONE 2013, 8, e70449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Chen, Z.; Shi, W.; Han, W.; Feng, Q.; Shi, C.; Engel, M.S.; Wang, S. Comparative Analysis of Complete Chloroplast Genomes of Nine Species of Litsea (Lauraceae): Hypervariable Regions, Positive Selection, and Phylogenetic Relationships. Genes 2022, 13, 1550. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Hilu, K.W.; Liang, G. The matK gene: Sequence variation and application in plant systematics. Am. J. Bot. 1997, 84, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Liao, R.; Yang, T.; Dong, X.; Lan, D.; Qin, R.; Liu, H. Aalysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). nBMC Genom. 2020, 21, 621. [Google Scholar] [CrossRef]
- Niu, Y.; Gao, C.; Liu, J. Comparative analysis of the complete plastid genomes of Mangifera species and gene transfer between plastid and mitochondrial genomes. PeerJ 2021, 9, e10774. [Google Scholar] [CrossRef]
- Chen, C.H.; Zheng, Y.J.; Liu, S.A.; Zhong, Y.D.; Wu, Y.; Li, J.; Xu, L.-A.; Xu, M. The complete chloroplast genome of Cinnamomum camphora and its comparison with related Lauraceae species. PeerJ 2017, 5, e3820. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Ma, P.-F.; Li, D.-Z. High-Throughput Sequencing of Six Bamboo Chloroplast Genomes: Phylogenetic Implications for Temperate Woody Bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [Green Version]
- Spalik, K.; Downie, S.R. Intercontinental disjunctions in Cryptotaenia (Apiaceae, Oenantheae): An appraisal using molecular data. J. Biogeogr. 2007, 34, 2039–2054. [Google Scholar] [CrossRef]
- Du, Y.-P.; Bi, Y.; Yang, F.-P.; Zhang, M.-F.; Chen, X.-Q.; Xue, J.; Zhang, X.-H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef] [Green Version]
- Buermans, H.P.J.; Den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Ma, C.; Zhang, Q.; Li, L.; Chen, X.; Zeng, H.; Guo, D. Leading dimensions in absorptive root trait variation across 96 subtropical forest species. New Phytol. 2014, 203, 863–872. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category of Genes | Group of Genes | Name of Genes |
|---|---|---|
| RNA genes | Transfer RNA | trnH-GUG, trnK-UUU b, trnQ-UUG, trnS-GCU, trnG-UCC b, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-GCC, trnfM-CAU, trnS-GGA, trnT-UGU, trnL-UAA b, trnF-GAA, trnV-UAC b, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU, trnL-CAA (×2), trnV-GAC (×2), trnI-GAU b (×2), trnA-UGC b (×2), trnR-ACG (×2), trnN-GUU (×2), trnL-UAG |
| Ribosomal RNA | rrn23 (×2), rrn16 (×2), rrn5 (×2), rrn4.5(×2) | |
| Transcription and translation related genes | DNA dependent RNA polymerase | rpoA, rpoB, rpoC1 b, rpoC2 |
| Large subunit of ribosome | rpl2 b, rpl14, rpl16 b, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12 ac (×2), rps14, rps15, rps16 b, rps18, rps19 | |
| Photosynthesis-related genes | ATP synthase | atpA, atpB, atpE, atpF b, atpH, atpI |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbI, psbJ, psbK, psbM, psbN, psbT, psbZ | |
| Cytochrome b/f complex | petA, petB b, petD b, petG, petL, petN | |
| Large subunit of rubisco | rbcL | |
| NADH dehydrogenase | ndhA b, ndhB b (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Other genes | Translational initiation factor | infA |
| Acetyl-CoA carboxylase | accD | |
| Maturase | matK | |
| Protease | clpP a | |
| Envelop membrane protein | cemA | |
| c-type cytochrom synthesis gene | ccsA | |
| Unknown function | Conserved open reading frames | ycf1, ycf2, ycf3 a, ycf4 |
| Gene | Location | ExonI(bp) | IntronI(bp) | ExonII (bp) | IntronII (bp) | ExonIII (bp) |
|---|---|---|---|---|---|---|
| ndhA + | SSC | 553 | 1119 | 539 | ||
| atpF − | LSC | 145 | 725 | 410 | ||
| clpP − | LSC | 71 | 776 | 294 | 652 | 244 |
| petB + | LSC | 6 | 790 | 642 | ||
| petD + | LSC | 8 | 716 | 475 | ||
| rpoC1 − | LSC | 453 | 718 | 1620 | ||
| rps16 − | LSC | 40 | 852 | 230 | ||
| trnG-UCC + | LSC | 23 | 765 | 48 | ||
| rpl16 − | LSC | 9 | 976 | 396 | ||
| rpl2 − | LSC | 392 | 672 | 430 | ||
| trnK-UUU − | LSC | 37 | 2507 | 35 | ||
| trnL-UAA + | LSC | 35 | 479 | 50 | ||
| rps12 # | LSC | 114 | — | 232 | 536 | 26 |
| trnV-UAC- | LSC | 39 | 589 | 35 | ||
| ycf3 − | LSC | 124 | 734 | 230 | 730 | 153 |
| ndhB + | IR | 721 | 702 | 758 | ||
| ndhB − | IR | 721 | 702 | 758 | ||
| trnA-UGC + | IR | 38 | 798 | 35 | ||
| trnA-UGC − | IR | 38 | 798 | 35 | ||
| trnI-GAU + | IR | 37 | 945 | 35 | ||
| trnI-GAU − | IR | 37 | 945 | 35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, X.; Peng, J.; Yang, Y.; Xiong, B. The Complete Chloroplast Genome Sequence of Machilus chuanchienensis (Lauraceae): Genome Structure and Phylogenetic Analysis. Genes 2022, 13, 2402. https://doi.org/10.3390/genes13122402
Bai X, Peng J, Yang Y, Xiong B. The Complete Chloroplast Genome Sequence of Machilus chuanchienensis (Lauraceae): Genome Structure and Phylogenetic Analysis. Genes. 2022; 13(12):2402. https://doi.org/10.3390/genes13122402
Chicago/Turabian StyleBai, Xue, Juan Peng, Yongyi Yang, and Biao Xiong. 2022. "The Complete Chloroplast Genome Sequence of Machilus chuanchienensis (Lauraceae): Genome Structure and Phylogenetic Analysis" Genes 13, no. 12: 2402. https://doi.org/10.3390/genes13122402
APA StyleBai, X., Peng, J., Yang, Y., & Xiong, B. (2022). The Complete Chloroplast Genome Sequence of Machilus chuanchienensis (Lauraceae): Genome Structure and Phylogenetic Analysis. Genes, 13(12), 2402. https://doi.org/10.3390/genes13122402