
Complete Chloroplast Genome Sequence of Endangered Species in the Genus Opisthopappus C. Shih: Characterization, Species Identification, and Phylogenetic Relationships

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA Extraction, and Sequencing
2.2. Cp genome Assembly and Annotation
2.3. Repeat Element Analysis
2.4. Codon Usage and RNA-Editing Sites Analysis
2.5. Phylogenetic and Divergence Time Analysis
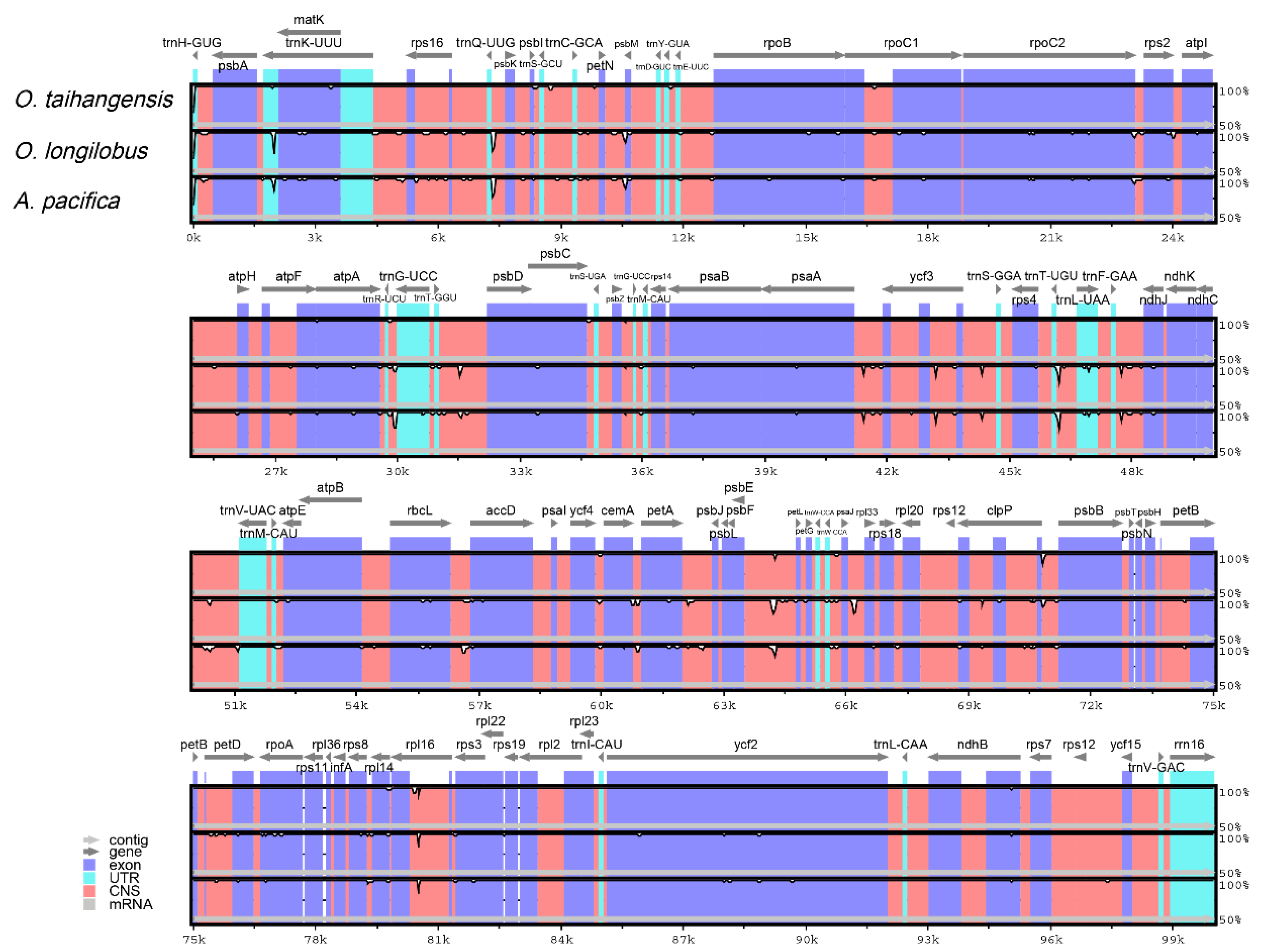
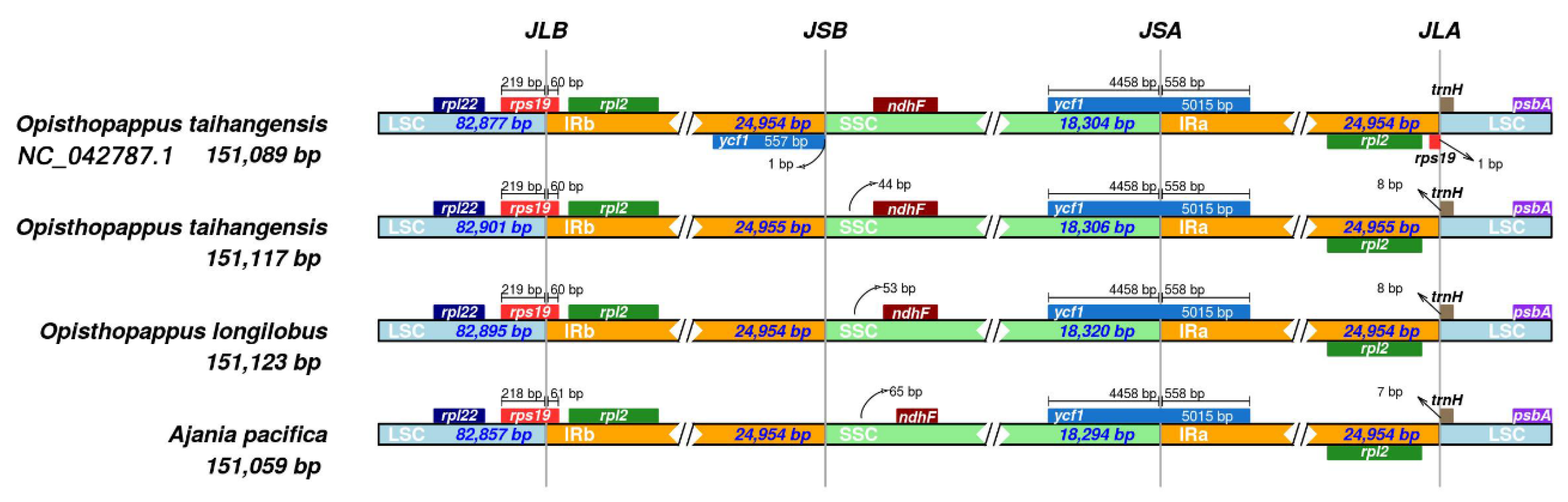
2.6. Genome Comparison
3. Results and Discussion
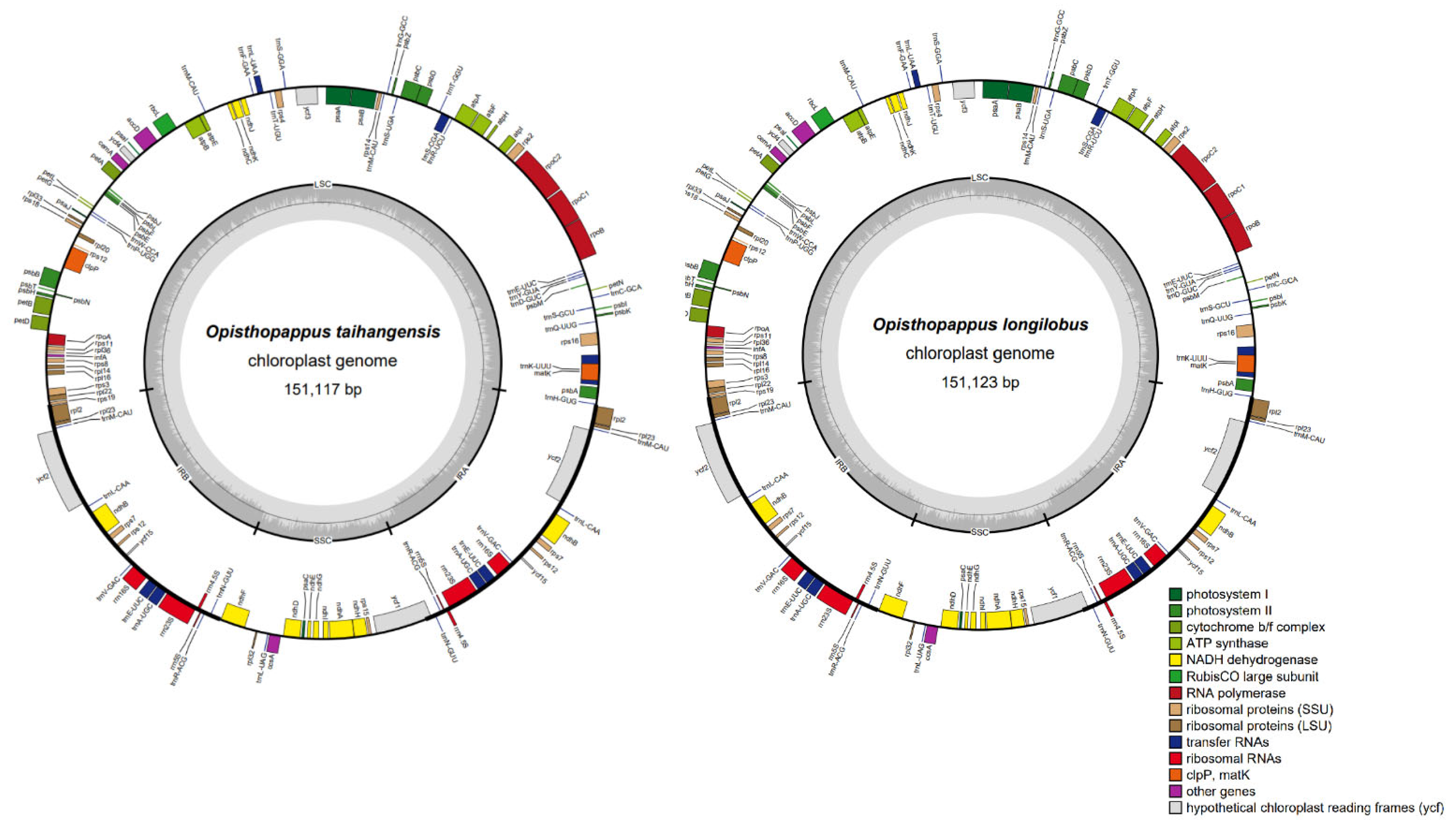
3.1. Characteristics of O. taihangensis and O. longilobus cp Genomes
3.2. Repetitive Sequence
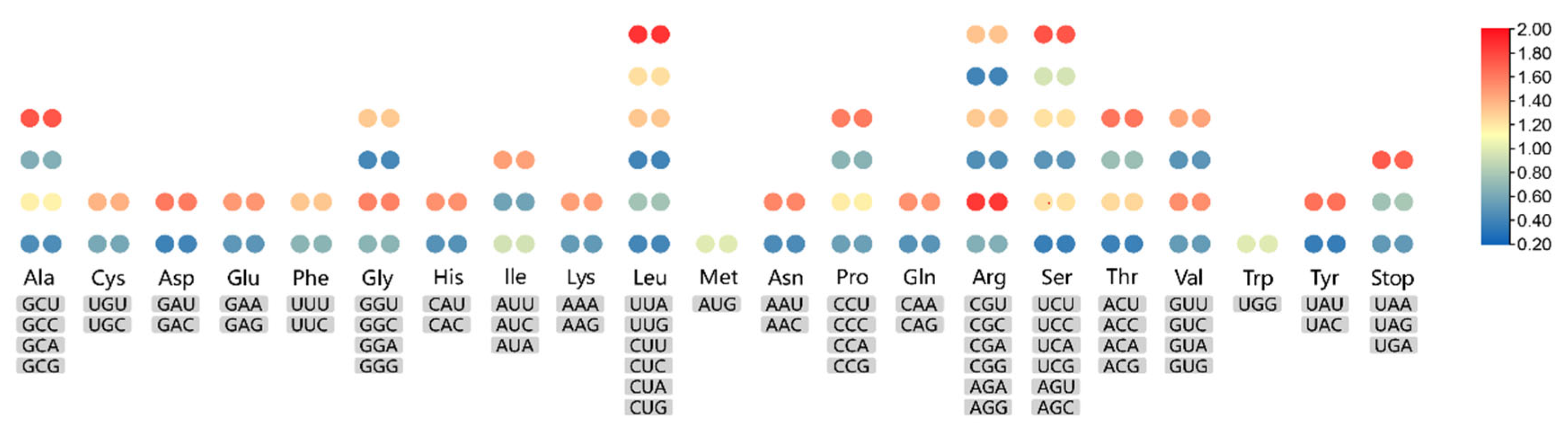
3.3. Codon Usage Analysis and RNA-Editing Sites
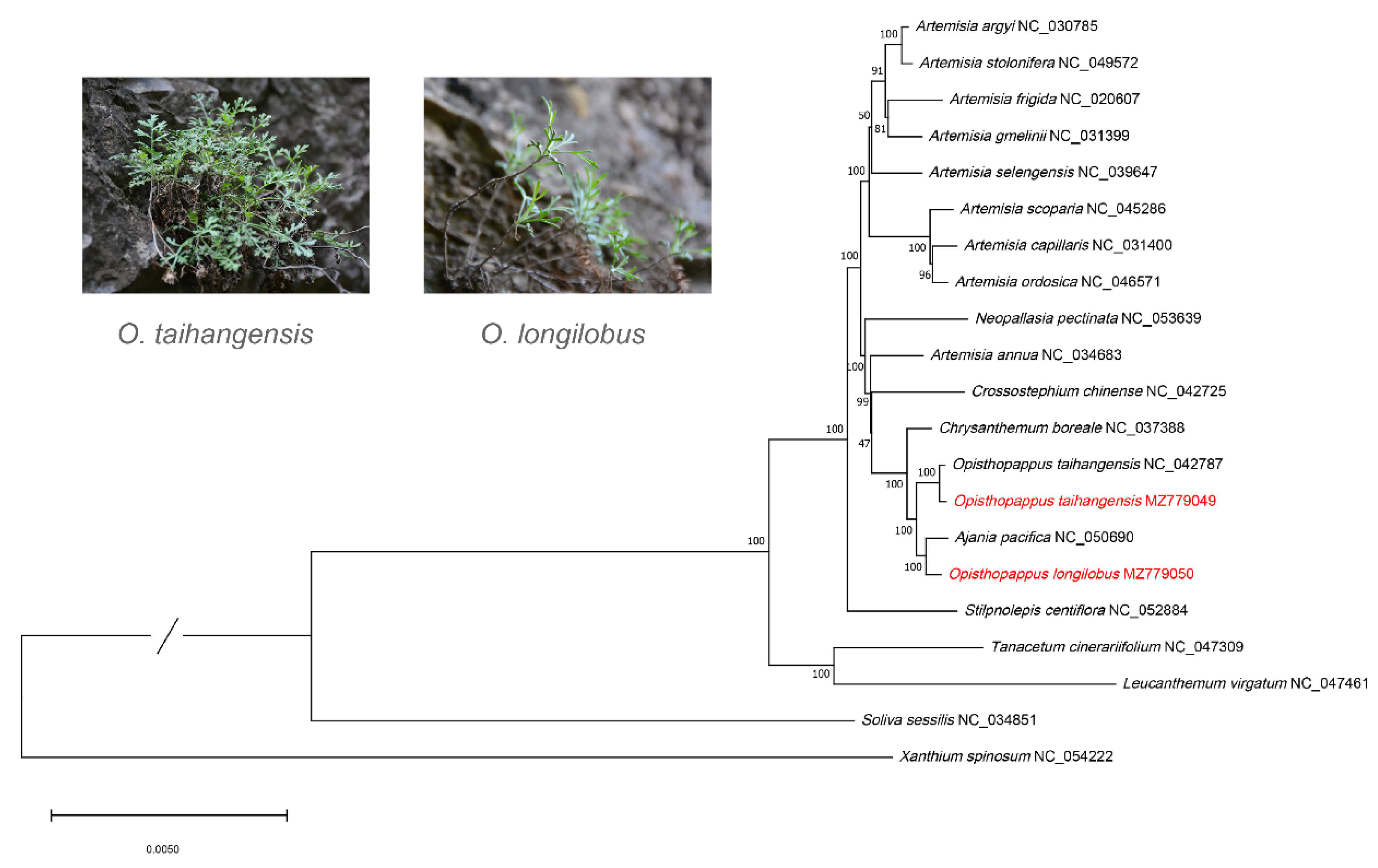
3.4. Phylogenetic Analysis and Divergence Time Analysis
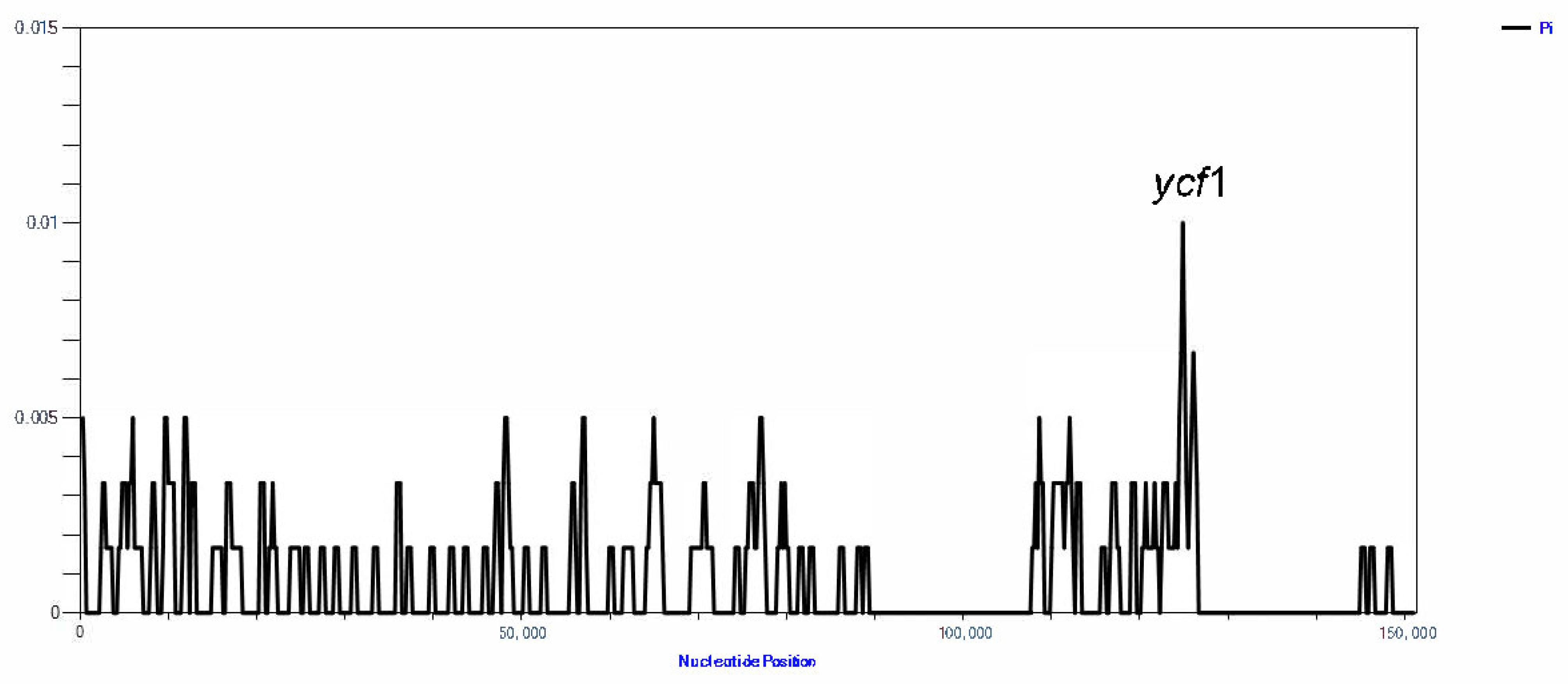
3.5. Comparative cp Genomic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| cp | chloroplast |
| NCBI | National Center for Biotechnology Information |
| LSC | large single-copy |
| SSC | small single-copy |
| IR | inverse repeat |
| SSRs | simple sequence repeats |
| ML | maximum likelihood |
| Mya | million years ago |
References
- Ye, H.; Wang, Z.; Hou, H.; Wu, J.; Gao, Y.; Han, W.; Ru, W.; Sun, G.; Wang, Y. Localized environmental heterogeneity drives the population differentiation of two endangered and endemic Opisthopappus Shih species. BMC Ecol. Evol. 2021, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-B.; Chen, F.-D.; Chen, S.-M.; Wu, G.-S.; Guo, W.-M. Molecular phylogeny of Chrysanthemum, Ajania and its allies (Anthemideae, Asteraceae) as inferred from nuclear ribosomal ITS and chloroplast trn LF IGS sequences. Plant Syst. Evol. 2010, 284, 153–169. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, X.-Q.; Ma, Y.-H. The Taihang and Yan mountains rose mainly in Quaternary. North China Earthq. Sci. 1999, 17, 1–7. [Google Scholar]
- Wei, D.; Xu, M.; Sun, W.; Jia, C.; Zhang, X. Antioxidant activity of aqueous extracts from different organs of Opisthopappus Shih. J. Chin. Inst. Food Sci. Technol. 2015, 15, 56–63. [Google Scholar]
- Chai, M.; Ye, H.; Wang, Z.; Zhou, Y.; Wu, J.; Gao, Y.; Han, W.; Zang, E.; Zhang, H.; Ru, W. Genetic divergence and relationship among Opisthopappus species identified by development of EST-SSR markers. Front. Genet. 2020, 11, 177. [Google Scholar] [CrossRef] [Green Version]
- Flora of China Editorial Committee. Flora of China; Missouri Botanical Garden and Harvard University Herbaria: St. Louis, MO, USA, 2018. [Google Scholar]
- Zhang, Z.; Xie, W.; Zhao, Y.; Zhang, J.; Wang, N.; Ntakirutimana, F.; Yan, J.; Wang, Y. EST-SSR marker development based on RNA-sequencing of E. sibiricus and its application for phylogenetic relationships analysis of seventeen Elymus species. BMC Plant Biol. 2019, 19, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Brunkard, J.O.; Runkel, A.M.; Zambryski, P.C. Chloroplasts extend stromules independently and in response to internal redox signals. Proc. Natl. Acad. Sci. USA 2015, 32, 10044–10049. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; De Pamphilis, C.W.; Kai, F.M.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What can we do with 1000 plastid genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Du, L.; Ao, L.; Chen, J.; Li, W.; Hu, W.; Zhang, W.; Kyunghee, K.; Sang-Choon, L.; Yang, T.J. The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Guo, S.; Xu, J.; He, L.; Carlson, J.E.; Hou, X. Phylogenetic analysis based on chloroplast genome uncover evolutionary relationship of all the nine species and six cultivars of tree peony. Ind. Crops Prod. 2020, 153, 112567. [Google Scholar] [CrossRef]
- Gu, J.; Wei, Z.; Yue, C.; Xu, X.; Zhang, T.; Zhao, Q.; Fu, S.; Yang, D.; Zhu, S. The complete chloroplast genome of Opisthopappus taihangensis (Ling) Shih. Mitochondrial DNA Part B 2019, 4, 1415–1416. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasnyanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, D.; Patrick, M.; Guillaume, S. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Stephan, G.; Pascal, L.; Ralph, B. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovli, I.; Zou, H.; Wang, G. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.-H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. PREP-Mt: Predictive RNA editor for plant mitochondrial genes. BMC Bioinform. 2005, 6, 1–15. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37 (Suppl. S2), W253–W259. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Frazer, K.A.; Lior, P.; Alexander, P.; Rubin, E.M.; Inna, D. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W. Many Parallel Losses of infA from Chloroplast DNA during Angiosperm Evolution with Multiple Independent Transfers to the Nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.; Yan, M.; Wang, J.; Zhao, L.; Diao, Y. The complete chloroplast genome sequences of five Miscanthus species, and comparative analyses with other grass plastomes. Ind. Crops Prod. 2021, 162, 113248. [Google Scholar] [CrossRef]
- Li, X.; Tan, W.; Sun, J.; Du, J.; Wang, Y. Comparison of Four Complete Chloroplast Genomes of Medicinal and Ornamental Meconopsis Species: Genome Organization and Species Discrimination. Sci. Rep. 2019, 9, 10567. [Google Scholar] [CrossRef]
- Irene, T.T.; Jordan, F.M.; Hawkins, J.A. Comparative phylogenetic methods and the cultural evolution of medicinal plant use. Nat. Plants 2018, 4, 754–761. [Google Scholar]
- Halim, S.; Prasad, R.S.; Dhobi, M. Quality Control Standardization and Comparative Anthelmintic Activity of Different Extracts from Evolvulus alsinoides L. Indian J. Pharm. Sci. 2020, 82, 356–361. [Google Scholar] [CrossRef]
- Wu, M.; Li, Q.; Hu, Z.; Li, X.; Chen, S. The Complete Amomum kravanh Chloroplast Genome Sequence and Phylogenetic Analysis of the Commelinids. Molecules 2017, 22, 1875. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| - | Features | O. taihangensis | O. longilobus |
|---|---|---|---|
| Accession Number | - | MZ779049 | MZ779050 |
| Length/bp | Total | 151,117 | 151,123 |
| LSC | 82,901 | 82,895 | |
| SSC | 18,306 | 18,320 | |
| IR | 24,955 | 24,954 | |
| GC content/% | Total | 37.46 | 37.44 |
| LSC | 35.54 | 35.52 | |
| SSC | 30.82 | 30.8 | |
| IR | 43.07 | 43.08 | |
| No. of gene | Total | 133 | 133 |
| Protein coding | 88 | 88 | |
| tRNA | 37 | 37 | |
| rRNA | 8 | 8 |
| Gene Function | Group Of Genes | Gene Names | Amount |
|---|---|---|---|
| rRNA | rRNA genes | rrn16S (×2), rrn23S (×2), rrn5S (×2), rrn4.5S (×2) | 8 |
| tRNA | tRNA genes | trnA-UGC (×2), trnC-GCA, trnD-GUC, trnE-UUC (×3), trnF-GAA, trnM-CAU (×4), trnG-GCC, trnH-GUG, trnK-UUU, trnL-CAA (×2), trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-CGA, trnS-GCU, trnS-GGA (×2), trnT-GGU, trnT-UGU, trnV-GAC(×2), trnW-CCA, trnY-GUA | 37 |
| Self-replication | Small subunit of ribosome | rps11, rps12 (×2), rps14, rps15 (×2), rps16, rps18, rps19, rps2, rps3, rps4, rps7 (×2), rps8 | 15 |
| Large subunit of ribosome | rpl14, rpl16, rpl2 (×2), rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 | 11 | |
| DNA dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | 4 | |
| Photosynthesis | Subunits of NADH-dehydrogenase | ndhA, ndhB(×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12 |
| Subunits of photosystem Ⅰ | psaA, psaB, psaC, psaI, psaJ | 5 | |
| Subunits of photosystem Ⅱ | psbA, psbB, psbC, psbD, psbE, psbF, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ, ycf3 | 15 | |
| Subunits of cytochrome b/f complex | petA, petB, petD, petG, petL, petN | 6 | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | 6 | |
| Large subunit of rubisco | rbcL | 1 | |
| Other genes | Maturase | matK | 1 |
| Protease | clpP | 1 | |
| Envelope membrane protein | cemA | 1 | |
| Subunit of Acetyl-CoA-carboxylase | accD | 1 | |
| c-type cytochrome synthesis gene | ccsA | 1 | |
| Translational initiation factor | infA | 1 | |
| Unknown | Conserved open reading frames | ycf1, ycf15(×2), ycf2 (×2), ycf4 | 6 |
| Total | 133 |
| Features | NC_042787 | MZ779049 | |
|---|---|---|---|
| Length/bp | 151089 | 151117 | |
| No. of gene | Total | 132 | 133 |
| Protein coding | 87 | 88 | |
| tRNA | 37 | 37 | |
| rRNA | 8 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhang, G.; Jiang, Y.; Huang, L. Complete Chloroplast Genome Sequence of Endangered Species in the Genus Opisthopappus C. Shih: Characterization, Species Identification, and Phylogenetic Relationships. Genes 2022, 13, 2410. https://doi.org/10.3390/genes13122410
Zhang X, Zhang G, Jiang Y, Huang L. Complete Chloroplast Genome Sequence of Endangered Species in the Genus Opisthopappus C. Shih: Characterization, Species Identification, and Phylogenetic Relationships. Genes. 2022; 13(12):2410. https://doi.org/10.3390/genes13122410
Chicago/Turabian StyleZhang, Xinke, Guoshuai Zhang, Yuan Jiang, and Linfang Huang. 2022. "Complete Chloroplast Genome Sequence of Endangered Species in the Genus Opisthopappus C. Shih: Characterization, Species Identification, and Phylogenetic Relationships" Genes 13, no. 12: 2410. https://doi.org/10.3390/genes13122410