
In Silico Prediction of Hub Genes Involved in Diabetic Kidney and COVID-19 Related Disease by Differential Gene Expression and Interactome Analysis

 ,
,
Abstract
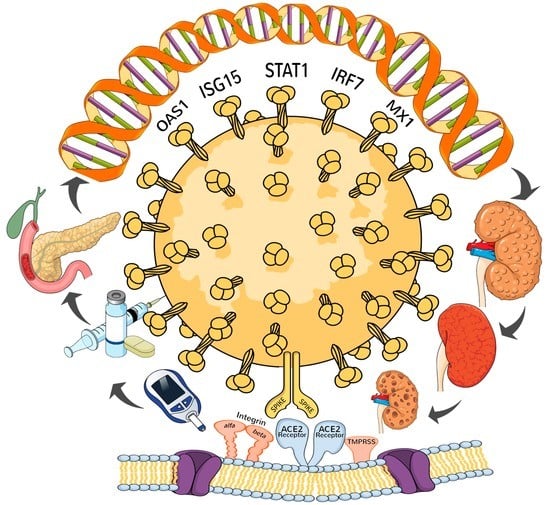
1. Introduction
2. Materials and Methods
2.1. Transcriptomic Data Acquisition
2.2. Differential Gene Expression Analysis
2.3. Functional Enrichment Analysis for DEGs in the DKD Dataset
2.4. Functional Enrichment Analysis for the Shared Pathogenic DEGs
2.5. PPI Network and Identification of Hub Genes
2.6. Deconvolution of the Gene Regulatory Network (GRN)
2.7. Functional Enrichment Analysis for Co-Expressed DEGs in the GRN
2.8. In Silico Validation of Expression of the Predictive Hub Genes in Coronascape DB
2.9. In Silico Validation of Expression of the Predictive Hub Genes in GTEx Database
3. Results
3.1. Identification of Differentially Expressed Genes across Diabetic Kidney Disease
3.2. DEGs in DKD Are Significantly Enriched in Cell and Immune Responses and COVID-19 Affected Pathways
3.3. Transcriptional Reprogramming of Gene Upregulation Is a Common Mechanism for DKD and COVID-19 Conditions
3.4. Interactome Analysis and Hub Genes Identification
3.5. In-Silico Validation of Hub Genes across COVID-19 and Healthy Tissue Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papatheodorou, K.; Banach, M.; Edmonds, M.; Papanas, N.; Papazoglou, D. Complications of diabetes. J. Diabetes Res. 2015, 2015, 189525. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Fadini, G.P. Microvascular complications in diabetes: A growing concern for cardiologists. Int. J. Cardiol. 2019, 291, 29–35. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Caramori, M.L.; Chan, J.C.; Heerspink, H.J.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A. KDIGO 2020 clinical practice guideline for diabetes management in chronic kidney disease. Kidney Int. 2020, 98, S1–S115. [Google Scholar] [CrossRef] [PubMed]
- Tye, S.C.; Denig, P.; Heerspink, H.J. Precision medicine approaches for diabetic kidney disease: Opportunities and challenges. Nephrol. Dial. Transpl. 2021, 36, ii3–ii9. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; Yu, T. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Deng, Q.; Feng, J.; Li, F.; Xiong, N.; He, Q. Clinical Characteristics of COVID-19 Patients with and without Diabetes in Wuhan Red Cross Hospital. J. Diabetes Res. 2020, 2020, 9756140. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Srivastava, R.; Chand, S.; Goodwin, J.E. Coronavirus disease COVID-19 and diabetic kidney disease. Pharmaceuticals 2021, 14, 751. [Google Scholar] [CrossRef]
- Leon-Abarca, J.A.; Memon, R.S.; Rehan, B.; Iftikhar, M.; Chatterjee, A. The impact of COVID-19 in diabetic kidney disease and chronic kidney disease: A population-based study. Acta Biomed. 2020, 91, e2020161. [Google Scholar] [CrossRef]
- Soltani, A.; Eskandari Mehrabadi, M.; Sheikhi, S.; Faghih, M.; Abdolmaleki, M. Routine Laboratory Parameters as a Tool for Predicting Death in Patients With COVID-19. Immunoregulation 2022, 4, 117–127. [Google Scholar] [CrossRef]
- Cruz, E.G.; Garcia, B.E.B.; Sandoval, D.M.; Gopar-Nieto, R.; Ruiz, F.J.G.; Gallardo, L.D.; Ronco, C.; Madero, M.; Jimenez, E.V. Renal Resistive Index as a Predictor of Acute Kidney Injury and Mortality in COVID-19 Critically Ill Patients. Blood Purif. 2022, 51, 309–316. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Inflammation in diabetic kidney disease. Nephron 2019, 143, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, J.; Zhu, G.; Zhang, Y.; Bi, Z.; Yu, Y.; Huang, B.; Fu, S.; Tan, Y.; Sun, J. Clinical features of maintenance hemodialysis patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. Clin. J. Am. Soc. Nephrol. 2020, 15, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Mourad, D.; Azar, N.S.; Azar, S.T. Diabetic nephropathy and COVID-19: The potential role of immune actors. Int. J. Mol. Sci. 2021, 22, 7762. [Google Scholar] [CrossRef] [PubMed]
- Ashish, K.; Idris, Y.; Ryan, W.; Madan, G.; Gaurav, G. COVID-19 in Kidney Transplantation: Epidemiology, Management Considerations, and the Impact on Kidney Transplant Practice. Transpl. Direct. 2020, 6, e582. [Google Scholar] [CrossRef]
- Muik, A.; Wallisch, A.; Snger, B.; Swanson, K.; Mühl, J.; Chen, W.; Cai, H.; Maurus, D.; Sarkar, R.; Türeci, Ö.; et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. Science 2021, 371, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Mallapaty, S.; Callaway, E. What scientists do and don’t know about the Oxford-AstraZeneca COVID vaccine. Nature 2021, 592, 15–17. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.d.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Li, Y.; Niu, L. Identification of the effects of COVID-19 on patients with pulmonary fibrosis and lung cancer: A bioinformatics analysis and literature review. Sci. Rep. 2022, 12, 1–17. [Google Scholar] [CrossRef]
- Koelle, K.; Martin, M.A.; Antia, R.; Lopman, B.; Dean, N.E. The changing epidemiology of SARS-CoV-2. Science 2022, 375, 1116–1121. [Google Scholar] [CrossRef]
- Jia, H.; Liu, C.; Li, D.; Huang, Q.; Liu, D.; Zhang, Y.; Ye, C.; Zhou, D.; Wang, Y.; Tan, Y. Metabolomic analyses reveal new stage-specific features of COVID-19. Eur. Respir. J. 2022, 59, 2100284. [Google Scholar] [CrossRef] [PubMed]
- Jahanafrooz, Z.; Chen, Z.; Bao, J.; Li, H.; Lipworth, L.; Guo, X. An overview of human proteins and genes involved in SARS-CoV-2 infection. Gene 2022, 808, 145963. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Lopez-Garcia, G.; Giorgi, F.M. corto: A lightweight R package for gene network inference and master regulator analysis. Bioinformatics 2020, 36, 3916–3917. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markie, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.; Tanaseichuk, O.; Christopher Benner, C.; Chanda, S. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Rydengård, V.; Olsson, A.K.; Mörgelin, M.; Schmidtchen, A. Histidine-rich glycoprotein exerts antibacterial activity. FEBS J. 2007, 274, 377–389. [Google Scholar] [CrossRef]
- Wuepper, K.D.; Miller, D.; Lacombe, M.J. Flaujeac trait. Deficiency of human plasma kininogen. J. Clin. Investig. 1975, 56, 1663–1672. [Google Scholar] [CrossRef]
- Rothschild, A.M.; Gomes, E.L.; Fortunato, I.C. Bradykinin release from high molecular weight kininogen and increase in plasma kallikrein-like activity following sensory stimulation by food in the rat. Naunyn Schmiedebergs Arch. Pharmacol. 1998, 358, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Aysert-Yildiz, P.; Ozger, H.S.; Yildiz, Y.; Buyukkoruk, M.; Yildiz, M.; Deveci-Bulut, T.S.; Gulbahar, O.; Dizbay, M. Prognostic Value of Serial Measurement of Serum Des-Arg (6)-Bradykinin Levels in Severe COVID-19 Patients. Clin. Lab. 2022, 68. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.L.; Stanisich, J.J.; Sauer, M.M.; Lin, C.-W.; Eras, J.; Zyla, D.S.; Trück, J.; Devuyst, O.; Aebi, M.; Pilhofer, M. Architecture and function of human uromodulin filaments in urinary tract infections. Science 2020, 369, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Woodruff, T.M. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J. Immunol. 2015, 194, 3542–3548. [Google Scholar] [CrossRef]
- Crozat, K.; Beutler, B. TLR7: A new sensor of viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6835–6836. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, Y.; Wang, X. CXCL10 an important chemokine associated with cytokine storm in COVID-19 infected patients. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7497–7505. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Valle, D.; Kim-Schulze, S.; Hsin-hui, H.; Beckmann, N.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.; Madduri, D.; Stock, A. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Banday, A.R.; Stanifer, M.L.; Florez-Vargas, O.; Onabajo, O.O.; Papenberg, B.W.; Zahoor, M.A.; Mirabello, L.; Ring, T.J.; Lee, C.-H.; Albert, P.S. Genetic regulation of OAS1 nonsense-mediated decay underlies association with COVID-19 hospitalization in patients of European and African ancestries. Nat. Genet. 2022, 54, 1103–1116. [Google Scholar] [CrossRef]
- Gibertoni, D.; Reno, C.; Rucci, P.; Fantini, M.P.; Buscaroli, A.; Mosconi, G.; Rigotti, A.; Giudicissi, A.; Mambelli, E.; Righini, M. COVID-19 incidence and mortality in non-dialysis chronic kidney disease patients. PLoS ONE 2021, 16, e0254525. [Google Scholar] [CrossRef] [PubMed]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5, e140711. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Ogawa, H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int. J. Hematol. 2021, 113, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Bagherimoghaddam, A.; Rafatpanah, H.; Mansouritorghabeh, H. Elevated levels of C3, C4, and CH50 of the complement system in ICU and non-ICU patients with COVID-19. Health Sci. Rep. 2022, 5, e519. [Google Scholar] [CrossRef] [PubMed]
- Leatherdale, A.; Stukas, S.; Lei, V.; West, H.E.; Campbell, C.J.; Hoiland, R.L.; Cooper, J.; Wellington, C.L.; Sekhon, M.S.; Pryzdial, E.L. Persistently elevated complement alternative pathway biomarkers in COVID-19 correlate with hypoxemia and predict in-hospital mortality. Med. Microbiol. Immunol. 2022, 211, 37–48. [Google Scholar] [CrossRef]
- Cheng, W.; Hornung, R.; Xu, K.; Li, J. Complement C3 identified as a unique risk factor for disease severity among young COVID-19 patients in Wuhan, China. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Ruffilli, I.; Ferrari, S.M.; Colaci, M.; Ferri, C.; Fallahi, P.; Antonelli, A. IP-10 in autoimmune thyroiditis. Horm. Metab. Res. 2014, 46, 597–602. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, C.; Li, J.; Yuan, J.; Yang, M.; Wang, F.; Li, G.; Li, Y.; Xing, L.; Peng, L. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J. Allergy Clin. Immunol. 2020, 146, 119–127.e114. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Burn, G.L.; Foti, A.; Marsman, G.; Patel, D.F.; Zychlinsky, A. The neutrophil. Immunity 2021, 54, 1377–1391. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147. [Google Scholar] [CrossRef] [PubMed]
- Anisul, M.; Shilts, J.; Schwartzentruber, J.; Hayhurst, J.; Buniello, A.; Mohammed, E.S.E.; Zheng, J.; Holmes, M.; Ochoa, D.; Carmona, M. A proteome-wide genetic investigation identifies several SARS-CoV-2-exploited host targets of clinical relevance. eLife 2021, 10, e69719. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Butler-Laporte, G.; Nakanishi, T.; Morrison, D.R.; Afilalo, J.; Afilalo, M.; Laurent, L.; Pietzner, M.; Kerrison, N.; Zhao, K. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med. 2021, 27, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-T.; Qiu, S.; Lv, L.-L.; Li, Z.-L.; Liu, H.; Tang, R.-N.; Ma, K.-L.; Liu, B.-C. Integrative bioinformatics analysis provides insight into the molecular mechanisms of chronic kidney disease. Kidney Blood Press. Res. 2018, 43, 568–581. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Xia, Y.; Yang, J. Systemic inflammation and chronic kidney disease in a patient due to the RNASEH2B defect. Pediatr. Rheumatol. 2021, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.; Gundersen, G.; Fernandez, N.; Wang, Z.; Monteiro, C.; McDermott, M.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Yasuda, S.; Kimura, T.; Nishio, S.; Kono, M.; Ohmura, K.; Shimamura, S.; Kono, M.; Fujieda, Y.; Kato, M. Interferon-inducible Mx1 protein is highly expressed in renal tissues from treatment-naïve lupus nephritis, but not in those under immunosuppressive treatment. Mod. Rheumatol. 2018, 28, 661–669. [Google Scholar] [CrossRef]
- Shyfrin, S.R.; Ferren, M.; Perrin-Cocon, L.; Espi, M.; Charmetant, X.; Brailly, M.; Decimo, D.; Iampietro, M.; Canus, L.; Horvat, B. Hamster organotypic kidney culture model of early-stage SARS-CoV-2 infection highlights a two-step renal susceptibility. J. Tissue Eng. 2022, 13, 20417314221122130. [Google Scholar] [CrossRef]
- Bai, J.; Wu, L.; Chen, X.; Wang, L.; Li, Q.; Zhang, Y.; Wu, J.; Cai, G.; Chen, X. Suppressor of cytokine signaling-1/STAT1 regulates renal inflammation in mesangial proliferative glomerulonephritis models. Front. Immunol. 2018, 9, 1982. [Google Scholar] [CrossRef]
- Rincon-Arevalo, H.; Aue, A.; Ritter, J.; Szelinski, F.; Khadzhynov, D.; Zickler, D.; Stefanski, L.; Lino, A.C.; Körper, S.; Eckardt, K.U. Altered increase in STAT1 expression and phosphorylation in severe COVID-19. Eur. J. Immunol. 2022, 52, 138–148. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, S.; Chou, J.; Faletti, L.E.; Haunerdinger, V.; Opitz, L.; Joset, P.; Fraser, C.J.; Prader, S.; Gao, X.; Schuch, L.A. Multisystem inflammation and susceptibility to viral infections in human ZNFX1 deficiency. J. Allergy Clin. Immunol. 2021, 148, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Maucourant, C.; Filipovic, I.; Ponzetta, A.; Aleman, S.; Cornillet, M.; Hertwig, L.; Strunz, B.; Lentini, A.; Reinius, B.; Brownlie, D. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 2020, 5, eabd6832. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Kretzschmar, D.; Yilmaz, A.; Bärthlein, B.; Titze, S.; Wolf, G.; Busch, M. Circulating dendritic cell precursors in chronic kidney disease: A cross-sectional study. BMC Nephrol. 2013, 14, 1–10. Available online: http://www.biomedcentral.com/1471-2369/14/274 (accessed on 5 October 2022). [CrossRef] [PubMed]
- Rogacev, K.S.; Seiler, S.; Zawada, A.M.; Reichart, B.; Herath, E.; Roth, D.; Ulrich, C.; Fliser, D.; Heine, G.H. CD14++ CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur. Heart J. 2011, 32, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Mou, W.; Su, C.; Chen, X.; Zhang, H.; Cao, B.; Li, X.; Wu, D.; Ni, X.; Gui, J. Increase in peripheral blood intermediate monocytes is associated with the development of recent-onset type 1 diabetes mellitus in children. Int. J. Biol. Sci. 2017, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Popescu, I.; Snyder, M.E.; Iasella, C.J.; Hannan, S.J.; Koshy, R.; Burke, R.; Das, A.; Brown, M.J.; Lyons, E.J.; Lieber, S.C. CD4+ T-Cell Dysfunction in Severe COVID-19 Disease Is Tumor Necrosis Factor-α/Tumor Necrosis Factor Receptor 1-Dependent. Am. J. Respir. Crit. Care Med. 2022, 205, 1403–1418. [Google Scholar] [CrossRef]
- Riou, C.; Du Bruyn, E.; Stek, C.; Daroowala, R.; Goliath, R.T.; Abrahams, F.; Said-Hartley, Q.; Allwood, B.W.; Hsiao, N.-Y.; Wilkinson, K.A. Relationship of SARS-CoV-2–specific CD4 response to COVID-19 severity and impact of HIV-1 and tuberculosis coinfection. J. Clin. Investig. 2021, 131, e149125. [Google Scholar] [CrossRef]
- Schroeder, S.; Pott, F.; Niemeyer, D.; Veith, T.; Richter, A.; Muth, D.; Goffinet, C.; Müller, M.A.; Drosten, C. Interferon antagonism by SARS-CoV-2: A functional study using reverse genetics. Lancet Microbe 2021, 2, e210–e218. [Google Scholar] [CrossRef]
- Xia, H.; Shi, P.-Y. Antagonism of type I interferon by severe acute respiratory syndrome coronavirus 2. J. Interf. Cytokine Res. 2020, 40, 543–548. [Google Scholar] [CrossRef]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Description | Gene Symbol | KEGG | Reactome |
|---|---|---|---|
| Complement C3 | C3 | * | NA |
| Caspase 1 | CASP1 | * | NA |
| C-C motif chemokine ligand 2 | CCL2 | * | NA |
| Complement factor B | CFB | * | * |
| C-X-C motif chemokine ligand 10 | CXCL10 | * | NA |
| C-X-C motif chemokine ligand 8 | CXCL8 | * | NA |
| Interferon α and β receptor subunit 2 | IFNAR2 | * | * |
| Interferon regulatory factor 7 | IRF7 | * | * |
| ISG15 ubiquitin like modifier | ISG15 | * | * |
| Janus kinase 1 | JAK1 | * | * |
| MX dynamin like GTPase 1 | MX1 | * | NA |
| 2′-5′-oligoadenylate synthetase 1 | OAS1 | * | * |
| Signal transducer and activator of transcription 1 | STAT1 | * | * |
| Toll like receptor 2 | TLR2 | * | NA |
| Toll like receptor 7 | TLR7 | * | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osuna-Martinez, U.; Aviña-Padilla, K.; Olimon-Andalon, V.; Angulo-Rojo, C.; Guadron-Llanos, A.; Rivas-Ferreira, J.C.; Urrea, F.; Calderon-Zamora, L. In Silico Prediction of Hub Genes Involved in Diabetic Kidney and COVID-19 Related Disease by Differential Gene Expression and Interactome Analysis. Genes 2022, 13, 2412. https://doi.org/10.3390/genes13122412
Osuna-Martinez U, Aviña-Padilla K, Olimon-Andalon V, Angulo-Rojo C, Guadron-Llanos A, Rivas-Ferreira JC, Urrea F, Calderon-Zamora L. In Silico Prediction of Hub Genes Involved in Diabetic Kidney and COVID-19 Related Disease by Differential Gene Expression and Interactome Analysis. Genes. 2022; 13(12):2412. https://doi.org/10.3390/genes13122412
Chicago/Turabian StyleOsuna-Martinez, Ulises, Katia Aviña-Padilla, Vicente Olimon-Andalon, Carla Angulo-Rojo, Alma Guadron-Llanos, Jose Carlos Rivas-Ferreira, Francisco Urrea, and Loranda Calderon-Zamora. 2022. "In Silico Prediction of Hub Genes Involved in Diabetic Kidney and COVID-19 Related Disease by Differential Gene Expression and Interactome Analysis" Genes 13, no. 12: 2412. https://doi.org/10.3390/genes13122412
APA StyleOsuna-Martinez, U., Aviña-Padilla, K., Olimon-Andalon, V., Angulo-Rojo, C., Guadron-Llanos, A., Rivas-Ferreira, J. C., Urrea, F., & Calderon-Zamora, L. (2022). In Silico Prediction of Hub Genes Involved in Diabetic Kidney and COVID-19 Related Disease by Differential Gene Expression and Interactome Analysis. Genes, 13(12), 2412. https://doi.org/10.3390/genes13122412