
Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot



Abstract
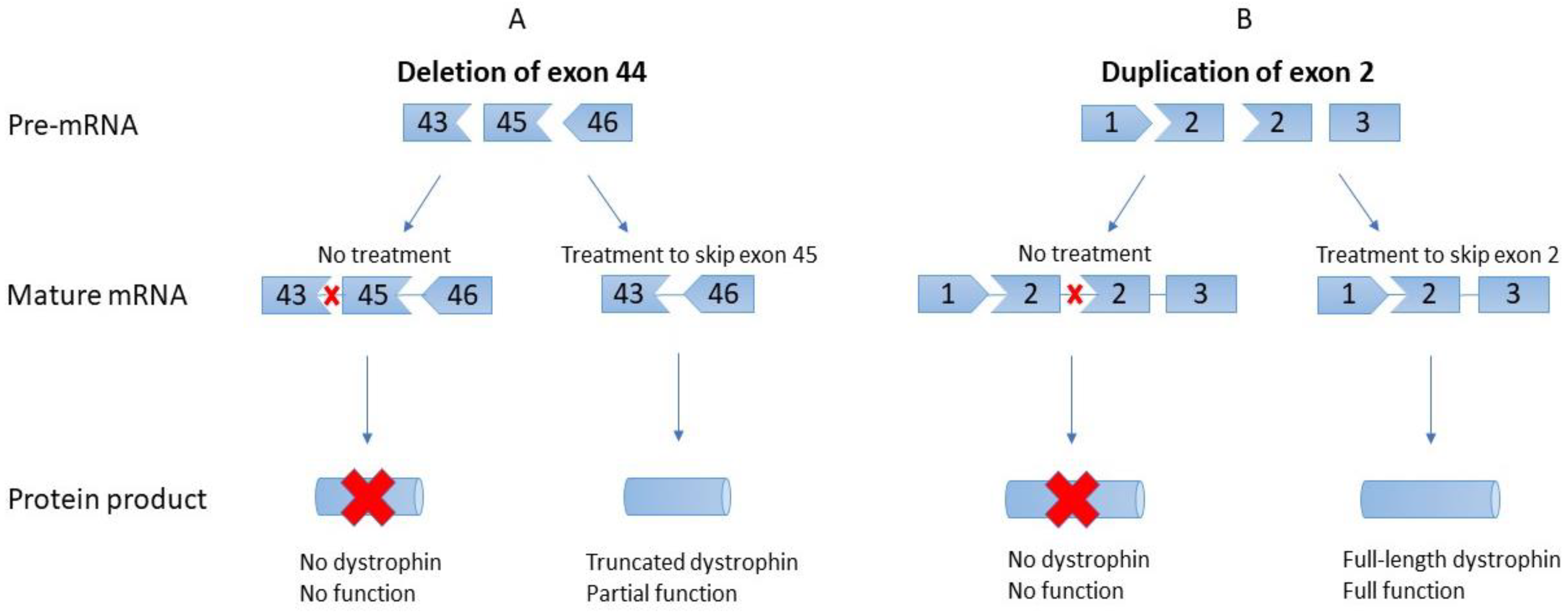
:1. Introduction
2. Exon 2 Skipping for Exon 2 Duplication—Astellas Gene Therapies
Strengths and Weaknesses
3. State of EST for Deletions
Strengths and Weaknesses
4. Generalized Gene Therapy
4.1. Pfizer-PF-06939926—Phase I
4.2. Pfizer-PF-06939926—Phase III
4.3. Sarepta-SRP-9001—Phase I
4.4. Sarepta-SRP-9001—Phase II
4.5. Sarepta-SRP-9001—ENDEAVOUR
4.6. Sarepta-SRP-9001—EMBARK
4.7. Solid Biosciences-SGT-001—IGNITE
4.8. Strengths and Weaknesses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet. J. Rare Dis. 2020, 15, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, V.; Pavlakis, S. Duchenne Muscular Dystrophy. StatPearls Publishing. 2021. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29493971 (accessed on 9 December 2021).
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne muscular dystrophy: From diagnosis to therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner-Medwin, D. Clinical features and classification of the muscular dystrophies. Br. Med. Bull. 1980, 36, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; De Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Rumeur, E.; Winder, S.J.; Hubert, J.F. Dystrophin: More than just the sum of its parts. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.J.; Davies, K.E. Duchenne muscular dystrophy and dystrophin: Pathogenesis and opportunities for treatment: Third in molecular medicine review series. EMBO Rep. 2004, 5, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Tennyson, C.N.; Klamut, H.J.; Worton, R.G. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat. Genet. 1995, 9, 184–190. [Google Scholar] [CrossRef]
- Mohammed, F.; Elshafey, A.; Al-balool, H.; Alaboud, H.; Al Ben Ali, M.; Baqer, A.; Bastaki, L. Mutation spectrum analysis of Duchenne/becker muscular dystrophy in 68 families in kuwait: The era of personalized medicine. PLoS ONE 2018, 13, e0197205. [Google Scholar] [CrossRef]
- Neri, M.; Rossi, R.; Trabanelli, C.; Mauro, A.; Selvatici, R.; Falzarano, M.S.; Spedicato, N.; Margutti, A.; Rimessi, P.; Fortunato, F.; et al. The genetic landscape of dystrophin mutations in italy: A nationwide study. Front. Genet. 2020, 11, 131. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple exon skipping in the Duchenne muscular dystrophy hot spots: Prospects and challenges. J. Pers. Med. 2018, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Gloss, D.; Moxley, R.T.; Ashwal, S.; Oskoui, M. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy—Report of the guideline development subcommittee of the american academy of neurology. Neurology 2016, 86, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Baker, D.E. Eteplirsen. Hosp. Pharm. 2017, 52, 302–305. [Google Scholar] [CrossRef]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today 2020, 56, 491–504. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Sinclair, A.; Islam, S.; Jones, S. Gene therapy: An overview of approved and pipeline technologies. In CADTH Issues in Emerging Health Technologies; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK538378/ (accessed on 13 December 2021).
- Scheller, E.L.; Krebsbach, P.H. Gene therapy: Design and prospects for craniofacial regeneration. J. Dent. Res. 2009, 88, 585. [Google Scholar] [CrossRef]
- Gonçalves, G.A.R.; Paiva, R.d.M.A. Gene therapy: Advances, challenges and perspectives. Einstein 2017, 15, 369. [Google Scholar] [CrossRef] [Green Version]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.R.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.A.; Yokota, T. Antisense therapy in neurology. J. Pers. Med. 2013, 3, 144–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lykken, E.A.; Shyng, C.; Edwards, R.J.; Rozenberg, A.; Gray, S.J. Recent progress and considerations for AAV gene therapies targeting the central nervous system. J. Neurodev. Disord. 2018, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-associated virus (AAV) as a vector for gene therapy. Biodrugs 2017, 31, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef]
- Brooks, P.J.; Ottinger, E.A.; Portero, D.; Lomash, R.M.; Alimardanov, A.; Terse, P.; Xu, X.; Chandler, R.J.; Hauserman, J.G.; Esposito, E.; et al. The platform vector gene therapies project: Increasing the efficiency of adeno-associated virus gene therapy clinical trial startup. Hum. Gene Ther. 2020, 31, 1034–1042. [Google Scholar] [CrossRef]
- Van Vliet, K.M.; Blouin, V.; Brument, N.; Agbandje-McKenna, M.; Snyder, R.O. The role of the adeno-associated virus capsid in gene transfer. Methods Mol. Biol. 2008, 437, 51–91. [Google Scholar]
- Berns, K.I.; Muzyczka, N. AAV: An overview of unanswered questions. Hum. Gene Ther. 2017, 28, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Pattali, R.; Mou, Y.; Li, X.J. AAV9 vector: A novel modality in gene therapy for spinal muscular atrophy. Gene Ther. 2019, 26, 287–295. [Google Scholar] [CrossRef]
- Wein, N.; Dunn, D.M.; Waldrop, M.A.; Gushchina, L.V.; Frair, E.C.; Weiss, R.B.; Flanigan, K.M. Absence of significant off-target splicing variation with a u7snrna vector targeting DMD exon 2 duplications. Hum. Gene Ther. 2021, 32, 1346–1359. [Google Scholar] [CrossRef]
- Vulin, A.; Wein, N.; Simmons, T.R.; Rutherford, A.M.; Findlay, A.R.; Yurkoski, J.A.; Kaminoh, Y.; Flanigan, K.M. The first exon duplication mouse model of Duchenne muscular dystrophy: A tool for therapeutic development. Neuromuscul. Disord. 2015, 25, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Wein, N.; Vulin, A.; Falzarano, M.S.; Szigyarto, C.A.K.; Maiti, B.; Findlay, A.; Heller, K.N.; Uhlén, M.; Bakthavachalu, B.; Messina, S.; et al. Translation from a DMD exon 5 ires results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat. Med. 2014, 20, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanigan, K.; Wein, N.; Gushchina, L.; Waldrop, M.; Weiss, R.P. 140rna-seq shows an absence of off-target splicing effects in AAV9-u7snrna mediated skipping of DMD exon 2. Neuromuscul. Disord. 2019, 29, S89. [Google Scholar] [CrossRef]
- Gushchina, L.V.; Frair, E.C.; Rohan, N.; Bradley, A.J.; Simmons, T.R.; Chavan, H.D.; Chou, H.J.; Eggers, M.; Waldrop, M.A.; Wein, N.; et al. Lack of toxicity in nonhuman primates receiving clinically relevant doses of an AAV9.u7snrna vector designed to induce DMD exon 2 skipping. Hum. Gene Ther. 2021, 32, 882–894. [Google Scholar] [CrossRef] [PubMed]
- clinicaltrials.gov. AAV9 U7snRNA Gene Therapy to Treat Boys with DMD Exon 2 Duplications. Available online: https://clinicaltrials.gov/ct2/show/NCT04240314 (accessed on 9 December 2021).
- National Cancer Institute. Common Terminology Criteria for Adverse Events (Ctcae) Common Terminology Criteria for Adverse Events (Ctcae) v5.0. 2017. Available online: https://www.meddra.org/ (accessed on 21 December 2021).
- Qu, Y.; Liu, Y.; Noor, A.; Tran, J.; Li, R. Characteristics and advantages of adeno-associated virus vector-mediated gene therapy for neurodegenerative diseases. Neural. Regen. Res. 2019, 14, 931. [Google Scholar] [PubMed]
- Alazard-dany, N.; Nicolas, A.; Ploquin, A.; Greco, A.; Epstein, A.L.; Fraefel, C. Packaging of genomes greater than 5 kb in length into AAV5 capsids is not efficient using a common vector production protocol. Mol. Ther. 2009, 17, S39. [Google Scholar]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; François, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23. [Google Scholar] [CrossRef]
- Ronzitti, G.; Gross, D.A.; Mingozzi, F. Human immune responses to adeno-associated virus (AAV) vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef]
- Weber, T. Anti-AAV antibodies in AAV gene therapy: Current challenges and possible solutions. Front. Immunol. 2021, 12, 702. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briefing Document Food and Drug Administration (FDA). Cellular, Tissue, and Gene Therapies Advisory Committee (Ctgtac) Meeting #70—Toxicity Risks of Adeno-Associated Virus (AAV) Vectors for Gene Therapy; FDA: Silver Spring, MA, USA, 2021.
- clinicaltrials.gov. Gene Transfer Clinical Study in x-Linked Myotubular Myopathy. Available online: https://clinicaltrials.gov/ct2/show/NCT03199469 (accessed on 27 December 2021).
- Philippidis, A. After third death, audentes’ AT132 remains on clinical hold. Hum. Gene Ther. 2020, 31, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S. High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020, 38, 910. [Google Scholar]
- Fourth Boy Dies in Trial of Astellas Gene Therapy Candidate. Available online: https://www.genengnews.com/news/fourth-boy-dies-in-trial-of-astellas-gene-therapy-candidate/ (accessed on 27 December 2021).
- Philippidis, A. Fourth boy dies in clinical trial of astellas’ AT132. Hum. Gene Ther. 2021, 32, 1008–1010. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Leborgne, C.; Barbon, E.; Alexander, J.; Hanby, H.; Delignat, S.; Cohen, D.; Collaud, F.; Muraleetharan, S.; Lupo, D.; Silverberg, J. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. igg-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing anti-bodies. Nat. Med. 2020, 26, 1096–1101. [Google Scholar] [CrossRef]
- Deyle, D.R.; Russell, D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther. 2009, 11, 442. [Google Scholar]
- Corey, D.R.; Abrams, J.M. Morpholino antisense oligonucleotides: Tools for investigating vertebrate development. Genome Biol. 2001, 2, reviews1015.1. [Google Scholar] [CrossRef]
- Nan, Y.; Zhang, Y.J. Antisense phosphorodiamidate morpholino oligomers as novel antiviral compounds. Front. Microbiol. 2018, 9, 750. [Google Scholar] [CrossRef]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. [Google Scholar]
- Okubo, M.; Noguchi, S.; Hayashi, S.; Nakamura, H.; Komaki, H.; Matsuo, M.; Nishino, I. Exon skipping induced by nonsense/frameshift mutations in DMD gene results in becker muscular dystrophy. Hum. Genet. 2020, 139, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhia, R.; Mishra, A.; Patel, V. Manipulation of renal gene expression using oligonucleotides. Methods Cell Biol. 2019, 154, 109–120. [Google Scholar] [PubMed]
- Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S. Deletion of exons 3–9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.T.; Barthelemy, F.; Martin, A.S.; Douine, E.D.; Eskin, A.; Lucas, A.; Lavigne, J.; Peay, H.; Khanlou, N.; Sweeney, L.; et al. DMD genotype correlations from the Duchenne registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum. Mutat. 2018, 39, 1193–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muntoni, F.; Gobbi, P.; Sewry, C.; Sherratt, T.; Taylor, J.; Sandhu, S.K.; Abbs, S.; Roberts, R.; Hodgson, S.V.; Bobrow, M.; et al. Deletions in the 5’ region of dystrophin and resulting phenotypes. J. Med. Genet. 1994, 31, 843. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, tolerability, and efficacy of viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping: A phase 2 randomized clinical trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef]
- Echigoya, Y.; Aoki, Y.; Miskew, B.; Panesar, D.; Touznik, A.; Nagata, T.; Tanihata, J.; Nakamura, A.; Nagaraju, K.; Yokota, T. Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45–55 with a cocktail of vivo-morpholinos in mdx52 mice. Mol. Ther. Nucleic. Acids 2015, 4, e225. [Google Scholar] [CrossRef]
- Wagner, K.R.; Kuntz, N.L.; Koenig, E.; East, L.; Upadhyay, S.; Han, B.; Shieh, P.B. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve 2021, 64, 285–292. [Google Scholar] [CrossRef]
- Hwang, J.; Yokota, T. Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies. Expert reviews in molecular medicine. Expert Rev. Mol. Med. 2019, 21, e5. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Yokota, T. An overview of recent advances and clinical applications of exon skipping and splice modulation for muscular dystrophy and various genetic diseases. Methods Mol. Biol. 2018, 1828, 31–55. [Google Scholar] [PubMed]
- AT702, AT751 and AT753—Duchenne Muscular Dystrophy—Astellas Gene Therapies Therapeutics. Available online: https://www.astellasgenetherapies.com/Duchenne/ (accessed on 10 December 2021).
- Himič, V.; Davies, K.E. Evaluating the potential of novel genetic approaches for the treatment of Duchenne muscular dystrophy. Eur. J. Hum. Genet. 2021, 29, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545. [Google Scholar] [CrossRef] [PubMed]
- Echevarría, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef] [Green Version]
- Jirka, S.M.G.; ‘t Hoen, P.A.C.; Diaz Parillas, V.; Tanganyika-de Winter, C.L.; Verheul, R.C.; Aguilera, B.; de Visser, P.C.; Aartsma-Rus, A.M. Cyclic peptides to improve delivery and exon skipping of antisense oligonucleotides in a mouse model for Duchenne muscular dystrophy. Mol. Ther. 2018, 26, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Duan, D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [Green Version]
- Duan, D. Micro-dystrophin gene therapy goes systemic in Duchenne muscular dystrophy patients. Hum. Gene Ther. 2018, 29, 733–736. [Google Scholar] [CrossRef]
- Shin, J.H.; Pan, X.; Hakim, C.H.; Yang, H.T.; Yue, Y.; Zhang, K.; Terjung, R.L.; Duan, D. Microdystrophin ameliorates muscular dystrophy in the canine model of Duchenne muscular dystrophy. Mol. Ther. 2013, 21, 750. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Pan, X.; Hakim, C.H.; Kodippili, K.; Zhang, K.; Shin, J.H.; Yang, H.T.; McDonald, T.; Duan, D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 2015, 24, 5880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- clinicaltrials.gov. A Study to Evaluate the Safety and Tolerability of pf-06939926 Gene Therapy in Duchenne Muscular Dystrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT03362502 (accessed on 9 December 2021).
- Pfizer Inc. Pfizer’s New Phase 1b Results of Gene Therapy in Ambulatory Boys with Duchenne Muscular Dystrophy (DMD) Support Advancement into Pivotal Phase 3 Study. Available online: https://investors.pfizer.com/investor-news/press-release-details/2020/Pfizers-New-Phase-1b-Results-of-Gene-Therapy-in-Ambulatory-Boys-with-Duchenne-Muscular-Dystrophy-DMD-Support-Advancement-into-Pivotal-Phase-3-Study/default.aspx (accessed on 9 December 2021).
- Safety and Efficacy of pf-06939926 Gene Therapy in Boys with Duchenne Muscular Dystrophy: Update on Data from the Phase 1b Study | Mda Clinical & Scientific Conference 2022. Available online: https://mdaconference.org/index.php/node/1168 (accessed on 9 December 2021).
- clinicaltrials.gov. A Phase 3 Study to Evaluate the Safety and Efficacy of pf-06939926 for the Treatment of Duchenne Muscular Dystrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT04281485 (accessed on 9 December 2021).
- Pfizer Tightens DMD Patient Criteria after Serious Adverse Events Crop up in Phase 3 Gene Therapy Trial | Fiercebiotech. Available online: https://www.fiercebiotech.com/biotech/pfizer-tightening-Duchenne-muscular-dystrophy-phase-3-criteria-adverse-events (accessed on 27 December 2021).
- Fierce Biotech. Pfizer Reports Patient Death in Early-Stage Duchenne Gene Therapy Trial, Halts Enrollment. Available online: https://www.fiercebiotech.com/biotech/pfizer-reports-death-patient-Duchenne-trial-halts-enrolment (accessed on 27 December 2021).
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of systemic delivery of rAAVrh74.mhck7.micro-dystrophin in children with Duchenne muscular dystrophy: A nonrandomized controlled trial. JAMA Neurol. 2020, 77, 1. [Google Scholar] [CrossRef] [PubMed]
- clinicaltrials.gov. Systemic Gene Delivery Clinical Trial for Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT03375164 (accessed on 27 December 2021).
- clinicaltrials.gov. A Randomized, Double-Blind, Placebo-Controlled Study of SRP-9001 for Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT03769116 (accessed on 9 December 2021).
- Sarepta Therapeutics, Inc. Sarepta Therapeutics Announces Top-Line Results for Part 1 of Study 102 Evaluating SRP-9001, Its Investigational Gene Therapy for the Treatment of Duchenne Muscular Dystrophy. Available online: https://investorrelations.Sarepta.com/news-releases/news-release-details/Sarepta-therapeutics-announces-top-line-results-part-1-study-102 (accessed on 9 December 2021).
- Roche. Roche Enters Licensing Agreement with Sarepta Therapeutics to Improve the Lives of Patients Living with Duchenne Muscular Dystrophy. Available online: https://www.roche.com/media/releases/med-cor-2019-12-23.htm (accessed on 9 December 2021).
- clinicaltrials.gov. A Gene Transfer Therapy Study to Evaluate the Safety of and Expression from SRP-9001 in Participants with Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT04626674 (accessed on 9 December 2021).
- Zaidman, C.; Proud, C.; Mcdonald, C.; Giblin, K.; Collins, L.; Wang, S.; Upadhyay, S.; Lewis, S.; Malhotra, J.; Griffin, D.A.; et al. ENDEAVOR: A gene delivery study to evaluate the safety of and expression from SRP-9001 in Duchenne muscular dystrophy. Available online: https://investorrelations.sarepta.com/static-files/a674d68e-823c-43a4-b26c-e6bfc6a5a95b (accessed on 9 December 2021).
- Sarepta Therapeutics, Inc. Sarepta Therapeutics’ SRP-9001 Shows Sustained Functional Improvements in Multiple Studies of Patients with Duchenne. Available online: https://investorrelations.Sarepta.com/news-releases/news-release-details/Sarepta-therapeutics-SRP-9001-shows-sustained-functional (accessed on 9 December 2021).
- clinicaltrials.gov. A Gene Transfer Therapy Study to Evaluate the Safety and Efficacy of SRP-9001 in Participants with Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT05096221 (accessed on 9 December 2021).
- clinicaltrials.gov. Microdystrophin Gene Transfer Study in Adolescents and Children with DMD. Available online: https://clinicaltrials.gov/ct2/show/NCT03368742 (accessed on 9 December 2021).
- Boehler, J.F.; Ricotti, V.; Gonzalez, J.P.; Soustek-Kramer, M.; Such, L.; Brown, K.J.; Schneider, J.S.; Morris, C.A. Membrane recruitment of nNOSµ in microdystrophin gene transfer to enhance durability. Neuromuscul. Disord. 2019, 29, 735–741. [Google Scholar] [CrossRef]
- BioSpace. FDA Slaps Clinical Hold on Solid Bioscience DMD Gene Therapy Program. Available online: https://www.biospace.com/article/fda-slaps-clinical-hold-on-solid-bioscience-DMD-gene-therapy-program/ (accessed on 27 December 2021).
- BioSpace. FDA Slaps Second Clinical Hold on Solid Biosciences’ DMD Gene Therapy due to Adverse Event. Available online: https://www.biospace.com/article/fda-slaps-second-clinical-hold-on-solid-biosciences-DMD-gene-therapy-due-to-adverse-event/ (accessed on 27 December 2021).
- Solid Biosciences. Solid Biosciences Reports Efficacy and Safety Data from the Ongoing Ignite DMD Clinical Trial and Resumption of Patient Dosing in the 2E14 vg/kg Cohort. Available online: https://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-efficacy-and-safety-data-from-the-ongoing-ignite-DMD-clinical-trial-and-resumption-of-patient-dosing-in-the-2e14-vg-kg-cohort (accessed on 9 December 2021).
- Wong, C.H.; Li, D.; Wang, N.; Gruber, J.; Conti, R.; Lo, A.W.; Campbell, J.; Gerrits, C.; Gooch, K.; Kowal, S.; et al. Estimating the financial impact of gene therapy *. medRxiv 2020. [Google Scholar] [CrossRef]


{kind=link}
| Trial | Drug Candidate | Phase | Study Type | Primary Endpoint | Estimated or Actual Primary Completion Date |
|---|---|---|---|---|---|
| NCT03362502 | PF-06939926 (Pfizer) | 1 | Open-label dose escalation study | Adverse events | February 2022 |
| NCT04281485 | PF-06939926 (Pfizer) | 3 | Randomized double-blind placebo-controlled study | Clinical efficacy with NSAA score | February 2023 |
| NCT04626674 (ENDEAVOR) | SRP-9001 (Sarepta) | 1 | Open-label efficacy study | Change in microdystrophin expression | March 2022 |
| NCT03375164 | SRP-9001 (Sarepta) | 1/2 | Open label safety study | Adverse events | April 2023 |
| NCT03769116 | SRP-9001 (Sarepta) | 2 | Randomized double-blind placebo-controlled study | Clinical efficacy with NSAA score and change in microdystrophin expression | December 2020 |
| NCT05096221 (EMBARK) | SRP-9001 (Sarepta) | 3 | Randomized double-blind placebo-controlled study | Clinical efficacy with NSAA score | October 2023 |
| NCT03368742 | SGT-001 (Solid Biosciences) | 1/2 | Open-label dose escalation study for safety and efficacy | Adverse events, change in microdystrophin expression, and clinical abnormalities | December 2023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilton-Clark, H.; Yokota, T. Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot. Genes 2022, 13, 257. https://doi.org/10.3390/genes13020257
Wilton-Clark H, Yokota T. Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot. Genes. 2022; 13(2):257. https://doi.org/10.3390/genes13020257
Chicago/Turabian StyleWilton-Clark, Harry, and Toshifumi Yokota. 2022. "Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot" Genes 13, no. 2: 257. https://doi.org/10.3390/genes13020257
APA StyleWilton-Clark, H., & Yokota, T. (2022). Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot. Genes, 13(2), 257. https://doi.org/10.3390/genes13020257