
Comprehensive Transcriptome–Metabolome Analysis and Evaluation of the Dark_Pur Gene from Brassica juncea that Controls the Differential Regulation of Anthocyanins in Brassica rapa


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plants Materials
2.2. DNA Extraction, RNA Extraction, and Reverse Transcription
2.3. Plant Genetic Transformation
2.4. Sequence Analysis of the Candidate Gene and Its Promoter
2.5. Sample Collection and RNA Sequencing
2.6. Transcriptome Data Preparation
2.7. Differential Gene Expression Analysis
2.8. Gene Functional Annotation and Enrichment Analysis
2.9. Quantitative Real Time PCR
2.10. Sample Preparation and Metabolite Extraction
2.11. LC-MS/MS Analysis and Metabolite Profiling Analysis
3. Results
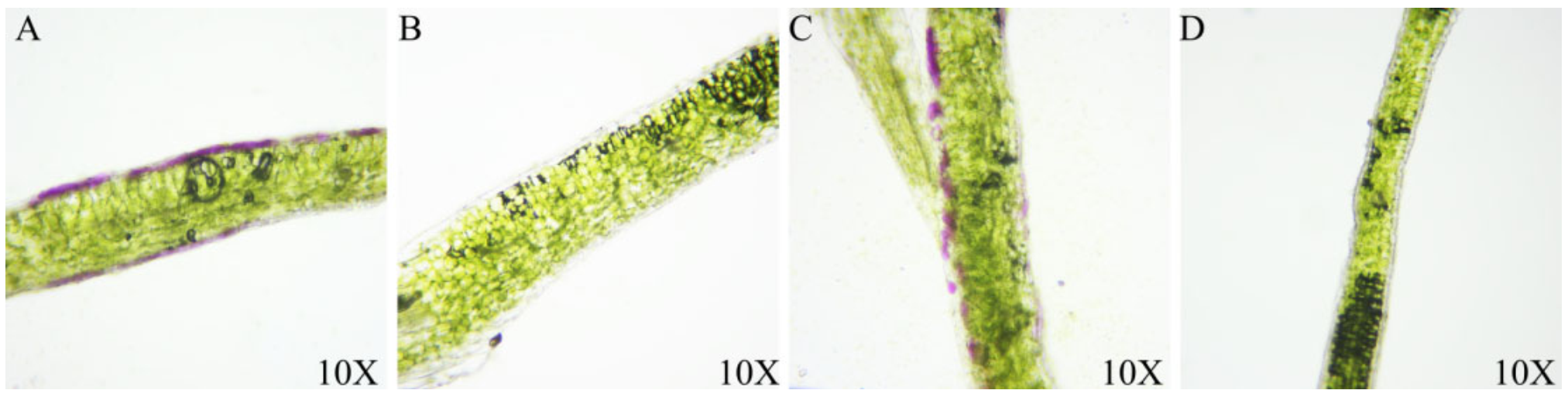
3.1. Purple Phenotype Analysis in B. rapa and B. juncea
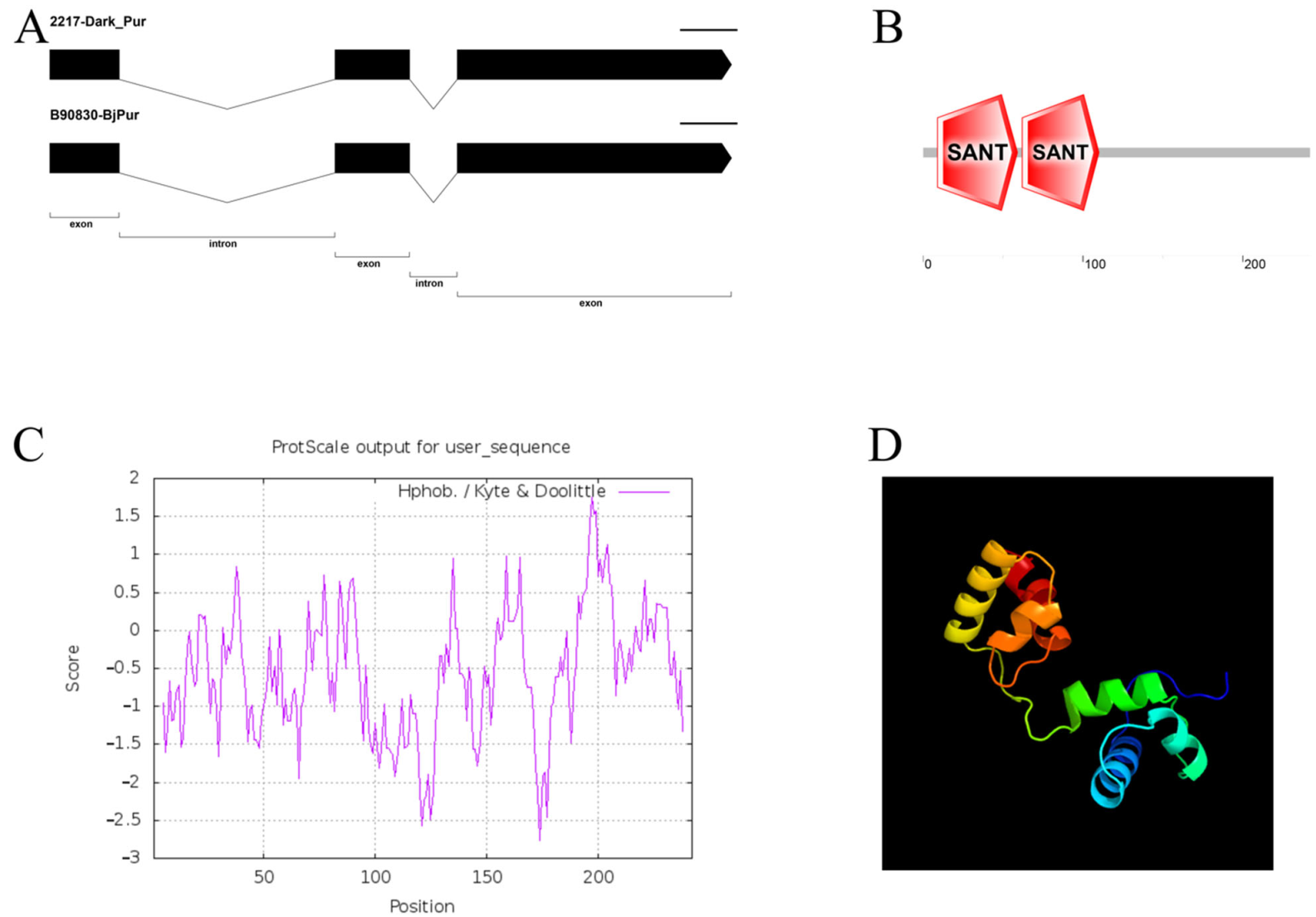
3.2. Cloning and Characteristic Analysis of the Dark_Pur Gene
3.3. Dark_Pur Gene Transformation in B. rapa for Functional Identification
3.4. Transcriptomic Analysis between Purple/Green B. rapa and B. juncea
3.4.1. Quality Control of the Transcriptomic Data
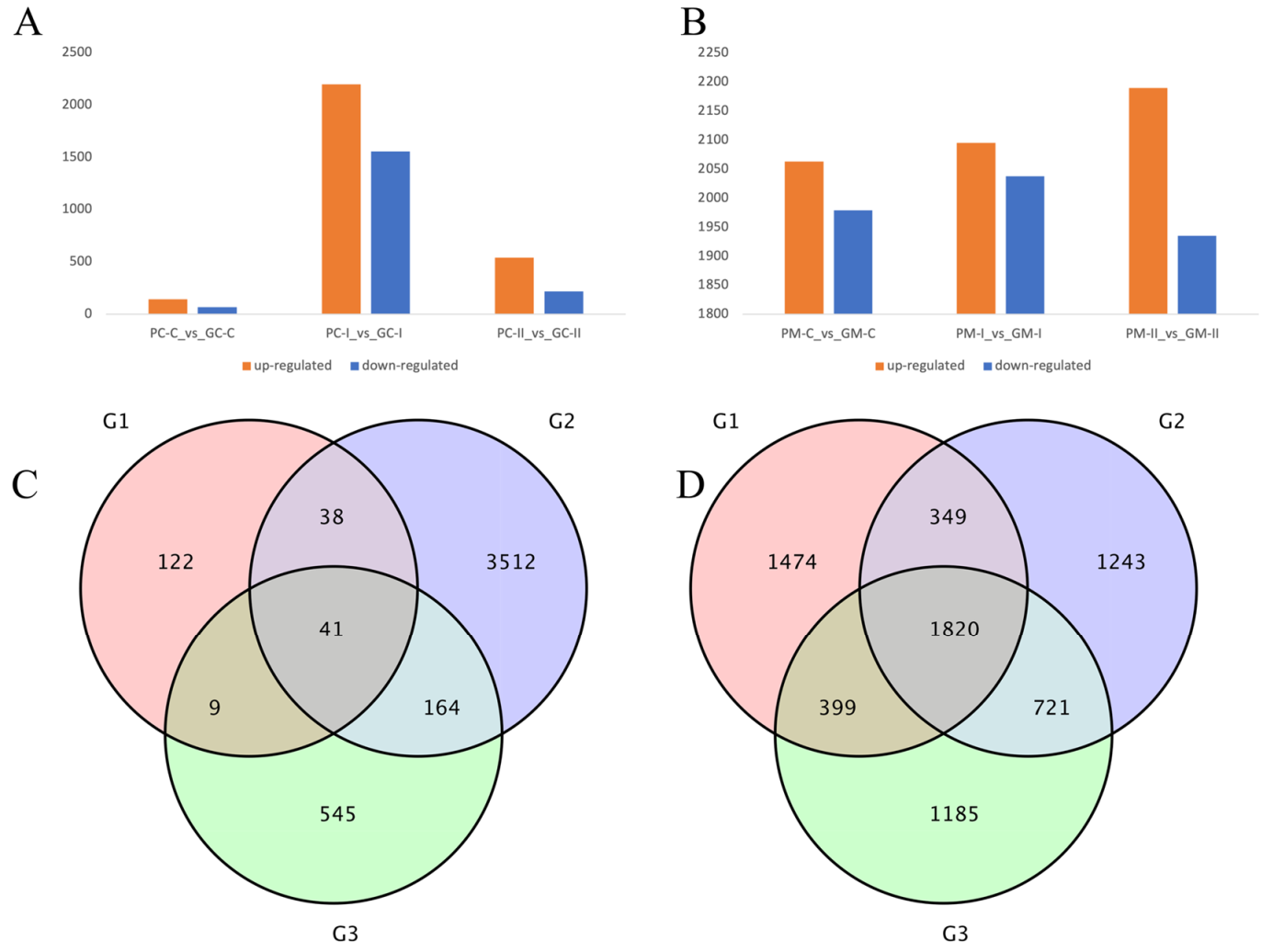
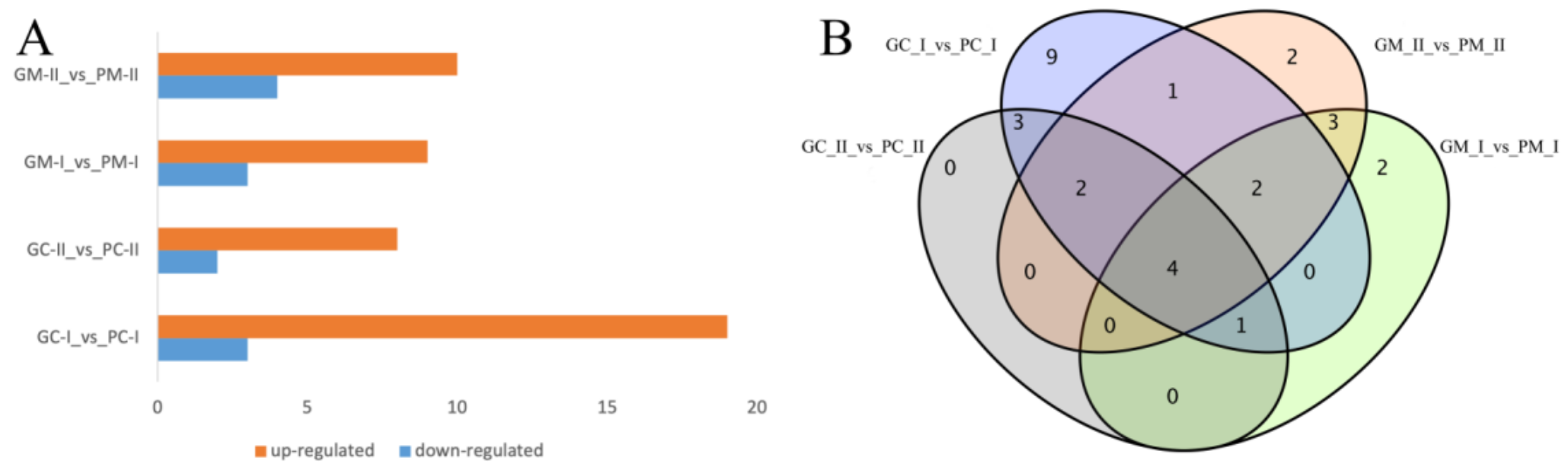
3.4.2. DEG Analysis between B. rapa and B. juncea
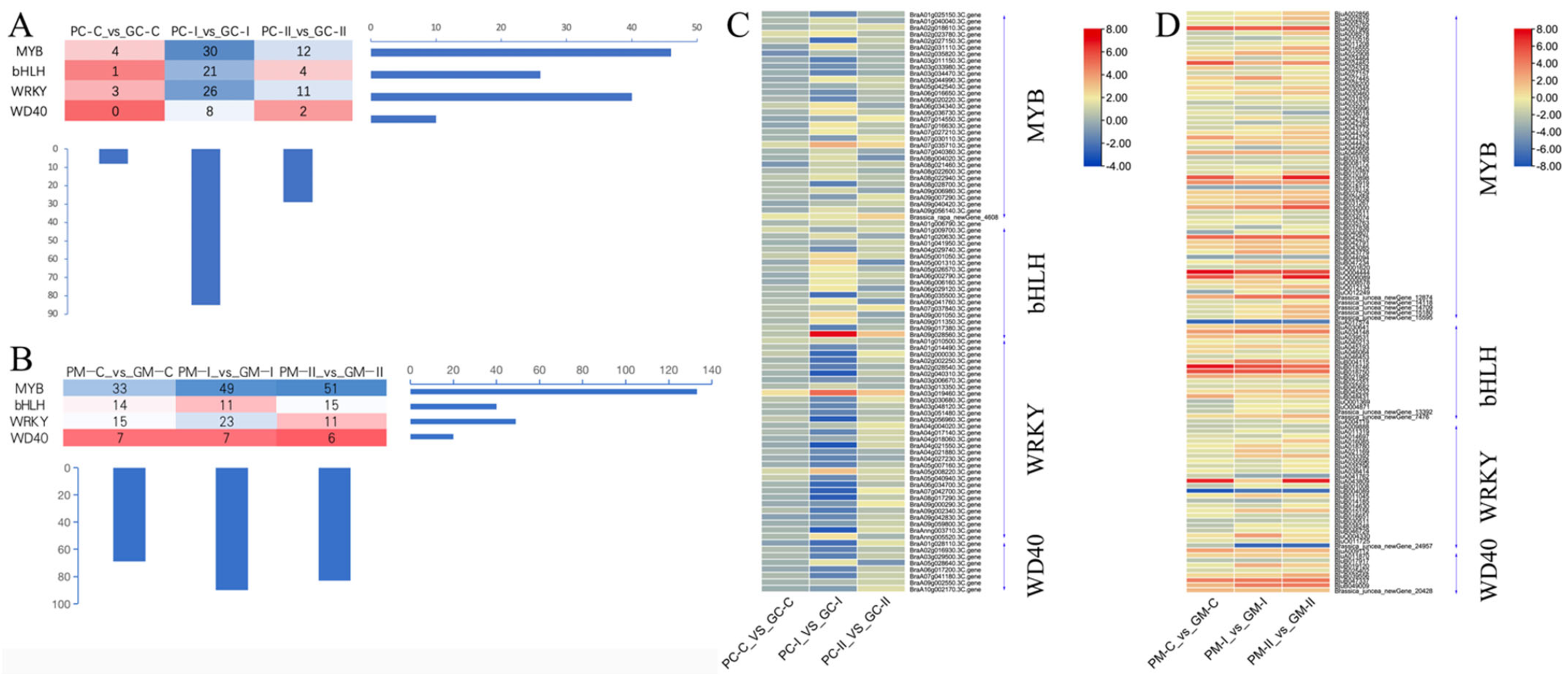
3.4.3. Transcription Factors Analysis of Anthocyanin Synthesis
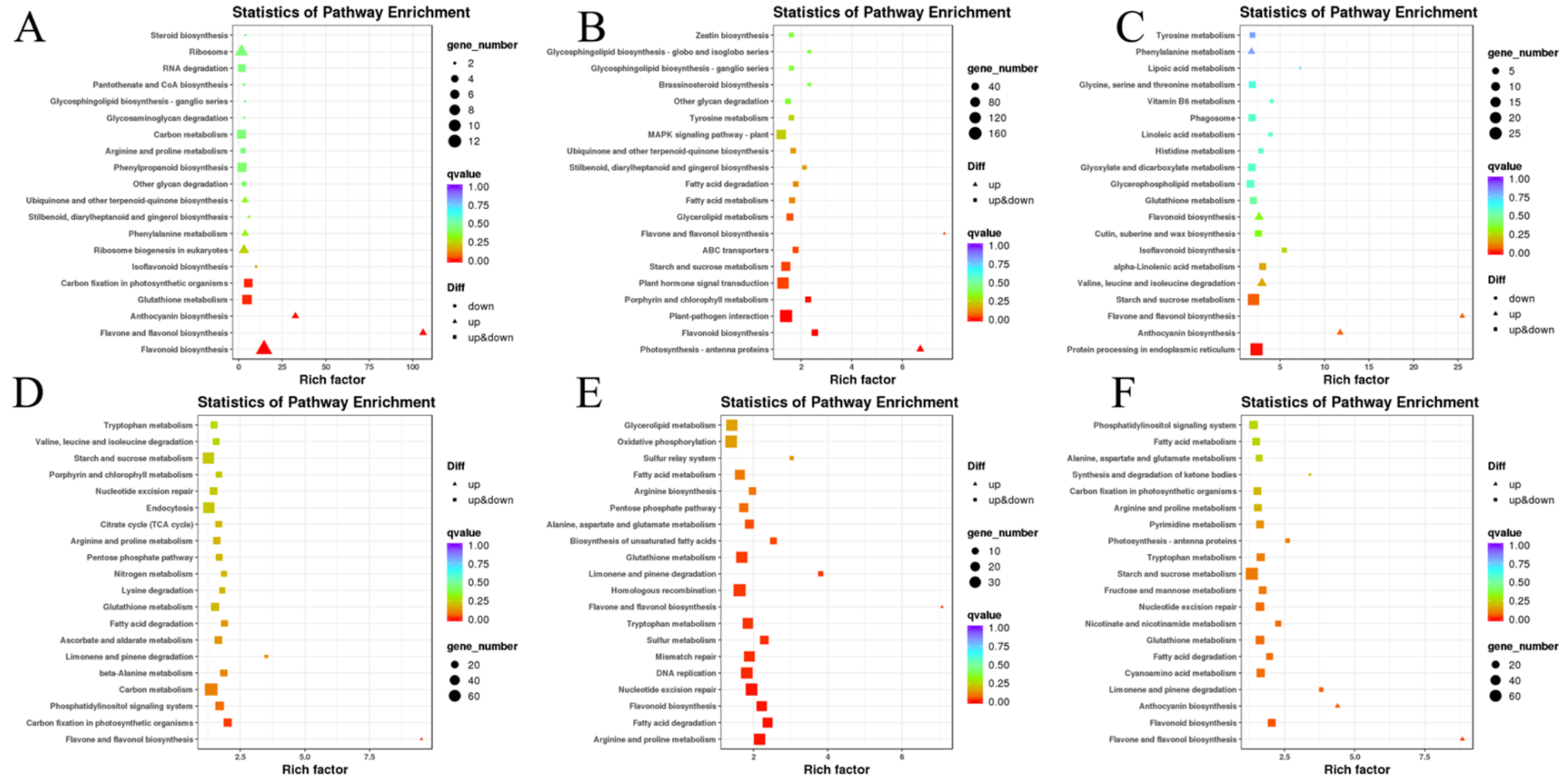
3.4.4. Gene Functional Annotation and Enrichment Analysis by GO and KEGG
3.5. The DEGs from the Transcriptome Analysis Were Detected by qRT-PCR
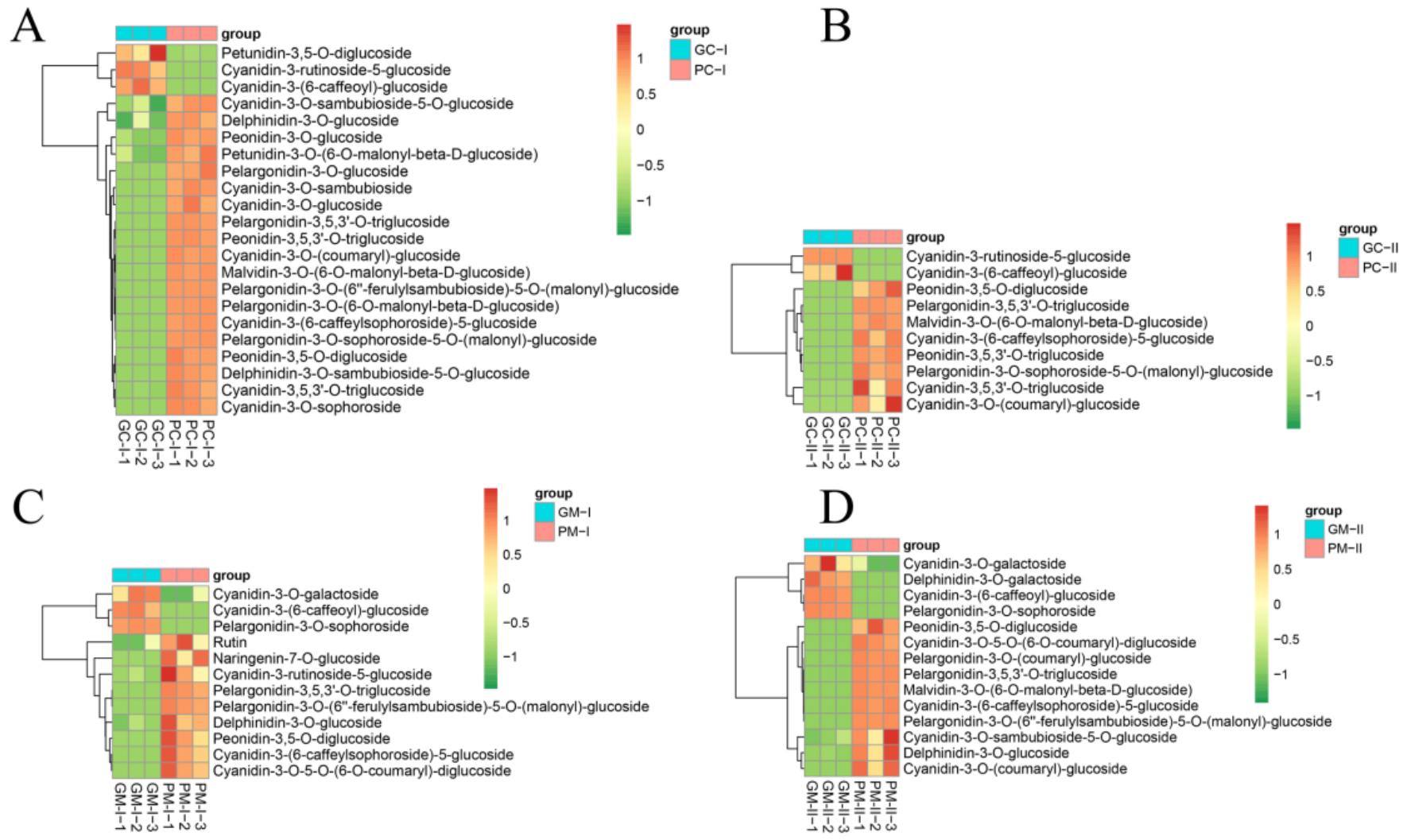
3.6. Metabolite Profiling Analysis between Purple/Green B. rapa and B. juncea
4. Discussion
4.1. The Structure Analysis and Transformation of the Dark_Pur Gene
4.2. Transcriptomic Analysis of B. rapa and B. juncea
4.3. The Expression Level of the Structure Genes of the B. rapa
4.4. KEGG Annotation of the B. rapa and B. juncea
4.5. Metabolome Analysis of the B. rapa and the B. juncea
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Castañeda-Ovando, A.; Pacheco-Hernandez, M.D.L.; Páez-Hernández, M.E.; Rodríguez, J.A.; Galán-Vidal, C.A. Chemical studies of anthocyanins: A review. Food Chem. 2009, 113, 859–871. [Google Scholar] [CrossRef]
- Hannum, S.M. Potential Impact of Strawberries on Human Health: A Review of the Science. Crit. Rev. Food Sci. Nutr. 2004, 44, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De Pascual-Teresa, S.; Moreno, D.A.; Garcia-Viguera, C. Flavanols and anthocyanins in cardiovascular health: A review of current evidence. Int. J. Mol. Sci. 2010, 11, 1679–1703. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-Induced Mutations in Grape Skin Color. Science 2004, 304, 982. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Lou, Q.; Liu, H.; Han, H.; Wang, Q.; Tang, Z.; Ma, Y.; Wang, H. Differential Regulation of Anthocyanins in Green and Purple Turnips Revealed by Combined De Novo Transcriptome and Metabolome Analysis. Int. J. Mol. Sci. 2019, 20, 4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, N.W.; Davies, K.M.; Lewis, D.H.; Zhang, H.; Montefiori, M.; Brendolise, C.; Boase, M.R.; Ngo, H.; Jameson, P.E.; Schwinn, K.E. A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in eudicots. Plant Cell 2014, 26, 962–980. [Google Scholar] [CrossRef] [Green Version]
- Ming, H.; Wang, Q.; Wu, Y.; Liu, H.; Zheng, L.; Zhang, G. Transcriptome analysis reveals the mechanism of anthocyanidins biosynthesis during grains development in purple corn (Zea mays L.). J. Plant Physiol. 2021, 257, 153328. [Google Scholar] [CrossRef]
- Zhao, J.; Dixon, R.A. The ‘ins’ and ‘outs’ of flavonoid transport. Trends Plant Sci. 2010, 15, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Gomez, C.; Conejero, G.; Torregrosa, L.; Cheynier, V.; Terrier, N.; Ageorges, A. In vivo grapevine anthocyanin transport involves vesicle-mediated trafficking and the contribution of anthoMATE transporters and GST. Plant J. 2011, 67, 960–970. [Google Scholar] [CrossRef]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the ttg1/bhlh/myb transcriptional complex in arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Lloyd, A.; Brockman, A.; Aguirre, L.; Campbell, A.; Bean, A.; Cantero, A.; Gonzalez, A. Advances in the myb-bhlh-wd repeat (mbw) pigment regulatory model: Addition of a wrky factor and co-option of an anthocyanin myb for betalain regulation. Plant Cell Physiol. 2017, 58, 1431–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Du, J.; Zhu, D.; Li, X.; Li, X. Metabolomic and transcriptomic analyses of anthocyanin biosynthesis mechanisms in the color mutant Ziziphus jujuba cv. Tailihong. J. Agric. Food Chem. 2020, 68, 15186–15198. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Xie, H.; Liu, S.; Zhu, J.; Zhao, S.; Wei, C. Metabolites and transcriptional profiling analysis reveal the molecular mechanisms of the anthocyanin metabolism in the “Zijuan” tea plant (Camellia sinensis var. Assamica). J. Agric. Food Chem. 2021, 69, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Cheng, F.; Wu, J.; Liu, B.; Zheng, S.; Liang, J.; Wang, X. Anthocyanin biosynthetic genes in Brassica rapa. BMC Genom. 2014, 15, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lin-Wang, K.; Deng, C.; Warran, B.; Wang, L.; Yu, B.; Yang, H.; Wang, J.; Espley, R.V.; Zhang, J.; et al. Comparative transcriptome analysis of white and purple potato to identify genes involved in anthocyanin biosynthesis. PLoS ONE 2015, 10, e0129148. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.H.; Kim, C.K. Roles of r2r3-myb transcription factors in transcriptional regulation of anthocyanin biosynthesis in horticultural plants. Plant Mol. Biol. 2018, 98, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Baudry, A.; Heim, M.A.; Dubreucq, B.; Caboche, M.; Weisshaar, B.; Lepiniec, L. Tt2, tt8, and ttg1 synergistically specify the expression of banyuls and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2004, 39, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Le Gourrierec, J.; Baudry, A.; Huep, G.; Lanet, E.; Debeaujon, I.; Routaboul, J.M.; Alboresi, A.; Weisshaar, B.; Lepiniec, L. Mybl2 is a new regulator of flavonoid biosynthesis in Arabidopsis thaliana. Plant J. 2008, 55, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Rubin, G.; Tohge, T.; Matsuda, F.; Saito, K.; Scheible, W.R. Members of the lbd family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 2009, 21, 3567–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, K.; Umemura, Y.; Ohme-Takagi, M. Atmybl2, a protein with a single myb domain, acts as a negative regulator of anthocyanin biosynthesis in arabidopsis. Plant J. 2008, 55, 954–967. [Google Scholar] [CrossRef]
- He, Q.; Ren, Y.; Zhao, W.; Li, R.; Zhang, L. Low temperature promotes anthocyanin biosynthesis and related gene expression in the seedlings of purple head chinese cabbage (Brassica rapa L.). Genes 2020, 11, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, L.W.; Zhou, X.; Burke, S.; Wu, X.; Prior, R.L.; Li, L. The purple cauliflower arises from activation of a myb transcription factor. Plant Physiol. 2010, 154, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, L.W.; Li, L. Characterization of the regulatory network of bomyb2 in controlling anthocyanin biosynthesis in purple cauliflower. Planta 2012, 236, 1153–1164. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Wu, J.; Guo, N.; Liang, J.; Wang, X.; Cheng, F. Qtl-seq and sequence assembly rapidly mapped the gene brmybl2.1 for the purple trait in Brassica rapa. Sci. Rep. 2020, 10, 2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, N.; Wu, J.; Zheng, S.; Cheng, F.; Liu, B.; Liang, J.; Cui, Y.; Wang, X. Anthocyanin profile characterization and quantitative trait locus mapping in zicaitai (Brassica rapa L. ssp. Chinensis var. Purpurea). Mol. Breed. 2015, 35, 113. [Google Scholar] [CrossRef]
- Li, G.H.; Chen, H.C.; Liu, J.L.; Luo, W.L.; Xie, D.S.; Luo, S.B.; Wu, T.Q.; Akram, W.; Zhong, Y.J. A high-density genetic map developed by specific-locus amplified fragment (slaf) sequencing and identification of a locus controlling anthocyanin pigmentation in stalk of zicaitai (Brassica rapa L. ssp. Chinensis var. Purpurea). BMC Genom. 2019, 20, 343. [Google Scholar] [CrossRef]
- Song, H.; Yi, H.; Lee, M.; Han, C.T.; Lee, J.; Kim, H.; Park, J.I.; Nou, I.S.; Kim, S.J.; Hur, Y. Purple Brassica oleracea var. capitata F. rubra is due to the loss of bomybl2-1 expression. BMC Plant Biol. 2018, 18, 82. [Google Scholar]
- Jin, S.-W.; Rahim, M.A.; Kim, H.-T.; Park, J.-I.; Kang, J.-G.; Nou, I.-S. Molecular analysis of anthocyanin-related genes in ornamental cabbage. Genome 2017, 61, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, G.; Dong, T.; Pan, Y.; Zhao, Z.; Tian, S.; Hu, Z. Anthocyanin accumulation and transcriptional regulation of anthocyanin biosynthesis in purple bok choy (Brassica rapa var. Chinensis). J. Agric. Food Chem. 2014, 62, 12366–12376. [Google Scholar] [CrossRef]
- Xie, L.; Li, F.; Zhang, S.; Zhang, H.; Qian, W.; Li, P.; Zhang, S.; Sun, R. Mining for candidate genes in an introgression line by using RNA sequencing: The anthocyanin overaccumulation phenotype in brassica. Front. Plant Sci. 2016, 7, 1245. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Wu, J.; Xue, Y.; Zhao, W.; Li, R.; Zhang, L. The novel gene brmyb2, located on chromosome a07, with a short intron 1 controls the purple-head trait of chinese cabbage (Brassica rapa L.). Hortic. Res. 2020, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xiao, L.; Xu, L.; Xing, X.; Tang, G.; Du, D. Fine mapping the bjpl1 gene for purple leaf color in b2 of brassica juncea l. Through comparative mapping and whole-genome re-sequencing. Euphytica 2017, 213, 80. [Google Scholar] [CrossRef]
- Heng, S.; Cheng, Q.; Zhang, T.; Liu, X.; Huang, H.; Yao, P.; Liu, Z.; Wan, Z.; Fu, T. Fine-mapping of the bjpur gene for purple leaf color in brassica juncea. Theor. Appl. Genet. 2020, 133, 2989–3000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, P.; Qian, W.; Zhang, S.; Li, F.; Zhang, H.; Wang, X.; Sun, R. Mapping and expression profiling reveal an inserted fragment from purple mustard involved anthocyanin accumulation in chinese cabbage. Euphytica 2016, 212, 83–95. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, J.; Qin, M.; Ren, Y.; Zhang, H.; Dai, Z.; Hao, L.; Zhang, L. Genetic analysis and mapping of the purple gene in purple heading chinese cabbage. Hortic. Plant J. 2016, 2, 351–356. [Google Scholar] [CrossRef]
- He, Q.; Zhang, Z.; Zhang, L. Anthocyanin accumulation, antioxidant ability and stability, and a transcriptional analysis of anthocyanin biosynthesis in purple heading chinese cabbage (Brassica rapa L. ssp. Pekinensis). J. Agric. Food Chem. 2016, 64, 132–145. [Google Scholar] [CrossRef]
- Zhang, L.; Cai, X.; Wu, J.; Liu, M.; Grob, S.; Cheng, F.; Liang, J.; Cai, C.; Liu, Z.; Liu, B.; et al. Improved brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 2018, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. Tbtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. Kegg for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the kegg orthology (ko) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Dai, Y.; Yuan, L.; Zhang, S.; Wang, J.; Xie, S.; Zhao, M.; Chen, G.; Sun, R.; Wang, C. Comprehensive evaluation for cold tolerance in wucai (Brassica campestris L.) by the performance index on an absorption basis (piabs). Agronomy 2019, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, T.; Horio, F.; Uchida, K.; Aoki, H.; Osawa, T. Dietary cyanidin 3-o-beta-d-glucoside-rich purple corn color prevents obesity and ameliorates hyperglycemia in mice. J. Nutr. 2003, 133, 2125–2130. [Google Scholar] [CrossRef]
- Zhang, Y.; Butelli, E.; Martin, C. Engineering anthocyanin biosynthesis in plants. Curr. Opin. Plant Biol. 2014, 19, 81–90. [Google Scholar] [CrossRef]
- Su, W.; Tao, R.; Liu, W.; Yu, C.; Yue, Z.; He, S.; Lavelle, D.; Zhang, W.; Zhang, L.; An, G.; et al. Characterization of four polymorphic genes controlling red leaf colour in lettuce that have undergone disruptive selection since domestication. Plant Biotechnol. J. 2020, 18, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Tian, Z.; Pan, Z.; Feng, X. Identification and quantification of anthocyanins in different coloured cultivars of ornamental kale (Brassica oleracea L. Var. acephala dc). J. Hortic. Sci. Biotechnol. 2017, 93, 466–473. [Google Scholar] [CrossRef]
- Xiang, L.; Liu, X.; Li, H.; Yin, X.; Grierson, D.; Li, F.; Chen, K. Cmmyb#7, an r3 myb transcription factor, acts as a negative regulator of anthocyanin biosynthesis in chrysanthemum. J. Exp. Bot. 2019, 70, 3111–3123. [Google Scholar]
- Zhong, C.; Tang, Y.; Pang, B.; Li, X.; Yang, Y.; Deng, J.; Feng, C.; Li, L.; Ren, G.; Wang, Y.; et al. The r2r3-myb transcription factor ghmyb1a regulates flavonol and anthocyanin accumulation in gerbera hybrida. Hortic. Res. 2020, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, Y.; Wang, H.; Tian, Z.; Xin, S.; Zhu, P. The dihydroflavonol 4-reductase bodfr1 drives anthocyanin accumulation in pink-leaved ornamental kale. Theor. Appl. Genet. 2021, 134, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Sun, H.; Sun, L.; Zhang, L. Transposon-induced methylation of the rsmyb1 promoter disturbs anthocyanin accumulation in red-fleshed radish. J. Exp. Bot. 2020, 71, 2537–2550. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Lu, Q.; He, Y.; Wang, Y.; Zhang, N.; Zhao, W.; Zhang, L. Dynamic changes of the anthocyanin biosynthesis mechanism during the development of heading chinese cabbage (Brassica rapa L.) and arabidopsis under the control of brmyb2. Front. Plant Sci. 2020, 11, 593766. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Su, T.B.; Yu, Y.J.; Zhang, D.S.; Wang, W.H.; Li, P.R.; Zhao, X.Y.; Yu, S.C.; Zhang, F.L. Molecular characterization of brmyb73: A candidate gene for the purple-leaf trait in Brassica rapa. Int. J. Agric. Biol. 2019, 22, 122–130. [Google Scholar]
- Merzlyak, M.N.; Solovchenko, A.E.; Chivkunova, O.B. Patterns of pigment changes in apple fruits during adaptation to high sunlight and sunscald development. Plant Physiol. Biochem. 2002, 40, 679–684. [Google Scholar] [CrossRef]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by myb-bhlh-wdr complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Jin, W.; Wang, H.; Li, M.; Wang, J.; Yang, Y.; Zhang, X.; Yan, G.; Zhang, H.; Liu, J.; Zhang, K. The r2r3 myb transcription factor pavmyb10.1 involves in anthocyanin biosynthesis and determines fruit skin colour in sweet cherry (Prunus avium L.). Plant Biotechnol. J. 2016, 14, 2120–2133. [Google Scholar] [CrossRef] [Green Version]
- Verweij, W.; Spelt, C.E.; Bliek, M.; de Vries, M.; Wit, N.; Faraco, M.; Koes, R.; Quattrocchio, F.M. Functionally similar wrky proteins regulate vacuolar acidification in petunia and hair development in arabidopsis. Plant Cell 2016, 28, 786–803. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, M.; Sakakibara, H.; Shinozaki, K.; et al. Enhancement of oxidative and drought tolerance in arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, I.M.; Heim, M.A.; Weisshaar, B.; Uhrig, J.F. Comprehensive identification of Arabidopsis thaliana myb transcription factors interacting with r/b-like bhlh proteins. Plant J. 2004, 40, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gonzalez, A.; Zhao, M.; Payne, C.T.; Lloyd, A. A network of redundant bhlh proteins functions in all ttg1-dependent pathways of Arabidopsis. Development 2003, 130, 4859–4869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, R.; Werber, M.; Weisshaar, B. The r2r3-myb gene family in arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Payne, C.; Zhang, F.; Lloyd, A. Gl3 encodes a bhlh protein that regulates trichome development in Arabidopsis through interaction with gl1 and ttg1. Genetics 2000, 156, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, L.; Hu, Z.; Chen, Y.; Tan, T.; Jia, Y.; Xie, Q.; Chen, G. Anthocyanin accumulation and transcriptional regulation of anthocyanin biosynthesis in purple pepper. J. Agric. Food Chem. 2020, 68, 12152–12163. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Li, G.; Zhang, S.; Zhang, S.; Zhang, H.; Sun, R.; Li, F. Comprehensive Transcriptome–Metabolome Analysis and Evaluation of the Dark_Pur Gene from Brassica juncea that Controls the Differential Regulation of Anthocyanins in Brassica rapa. Genes 2022, 13, 283. https://doi.org/10.3390/genes13020283
Liu Y, Li G, Zhang S, Zhang S, Zhang H, Sun R, Li F. Comprehensive Transcriptome–Metabolome Analysis and Evaluation of the Dark_Pur Gene from Brassica juncea that Controls the Differential Regulation of Anthocyanins in Brassica rapa. Genes. 2022; 13(2):283. https://doi.org/10.3390/genes13020283
Chicago/Turabian StyleLiu, Yujia, Guoliang Li, Shujiang Zhang, Shifan Zhang, Hui Zhang, Rifei Sun, and Fei Li. 2022. "Comprehensive Transcriptome–Metabolome Analysis and Evaluation of the Dark_Pur Gene from Brassica juncea that Controls the Differential Regulation of Anthocyanins in Brassica rapa" Genes 13, no. 2: 283. https://doi.org/10.3390/genes13020283
APA StyleLiu, Y., Li, G., Zhang, S., Zhang, S., Zhang, H., Sun, R., & Li, F. (2022). Comprehensive Transcriptome–Metabolome Analysis and Evaluation of the Dark_Pur Gene from Brassica juncea that Controls the Differential Regulation of Anthocyanins in Brassica rapa. Genes, 13(2), 283. https://doi.org/10.3390/genes13020283