
Phenotypic Variation in Vietnamese Osteogenesis Imperfecta Patients Sharing a Recessive P3H1 Pathogenic Variant

 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genomic DNA Extraction and Sanger Sequencing of COL1A1 and COL1A2
2.3. Targeted Next-Generation Sequencing
3. Results
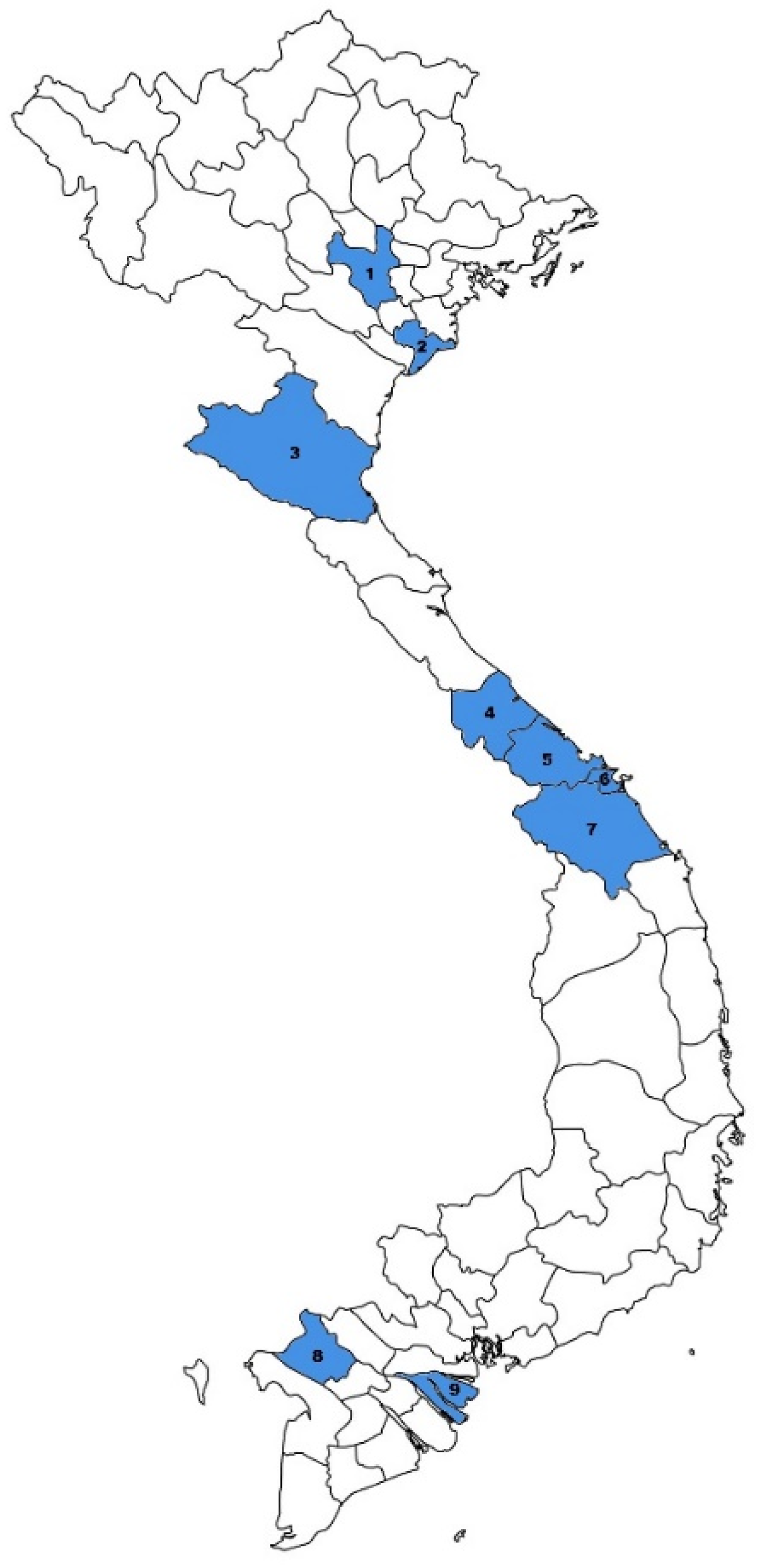
3.1. General Description of the Cohort
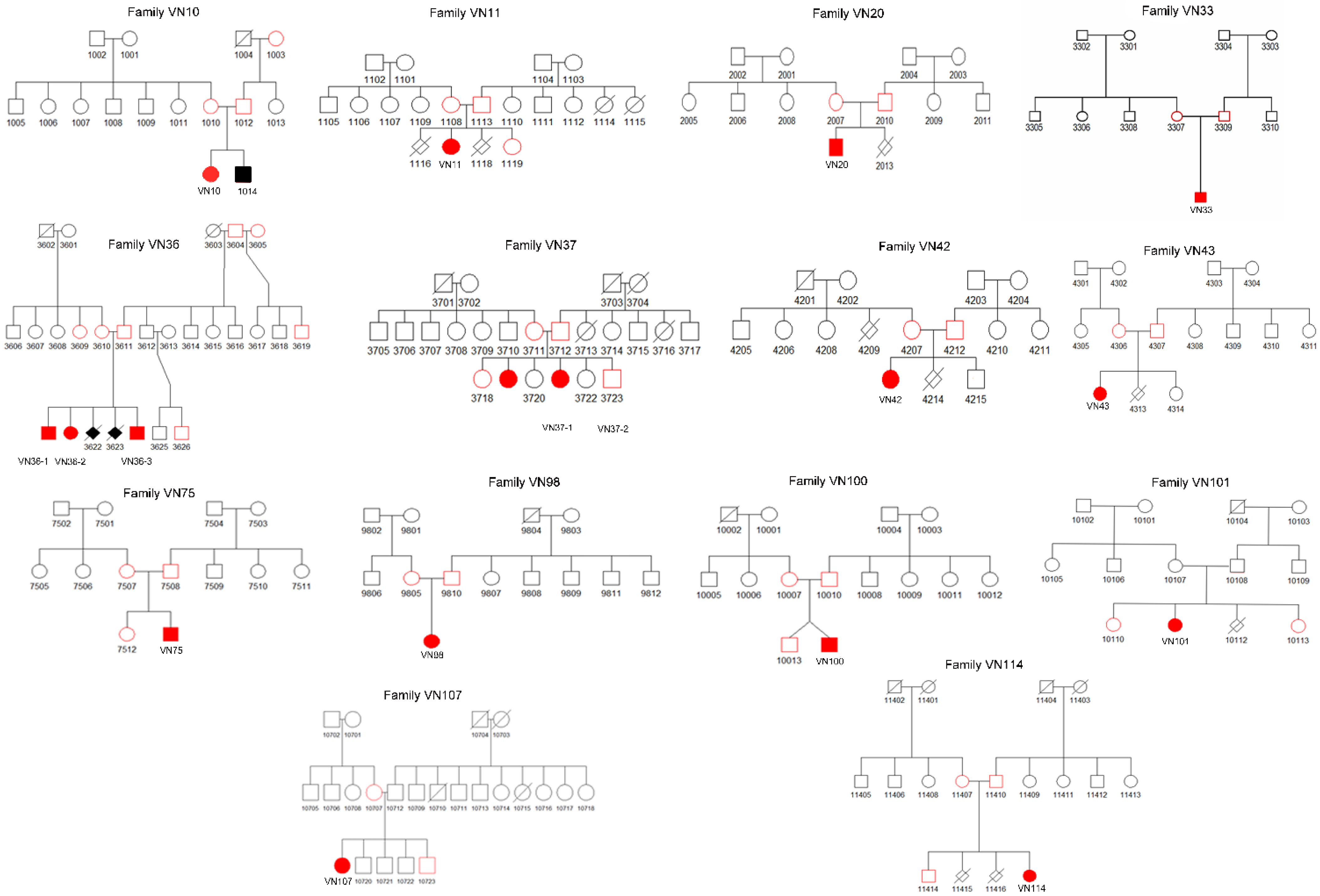
3.2. Genetic Analysis
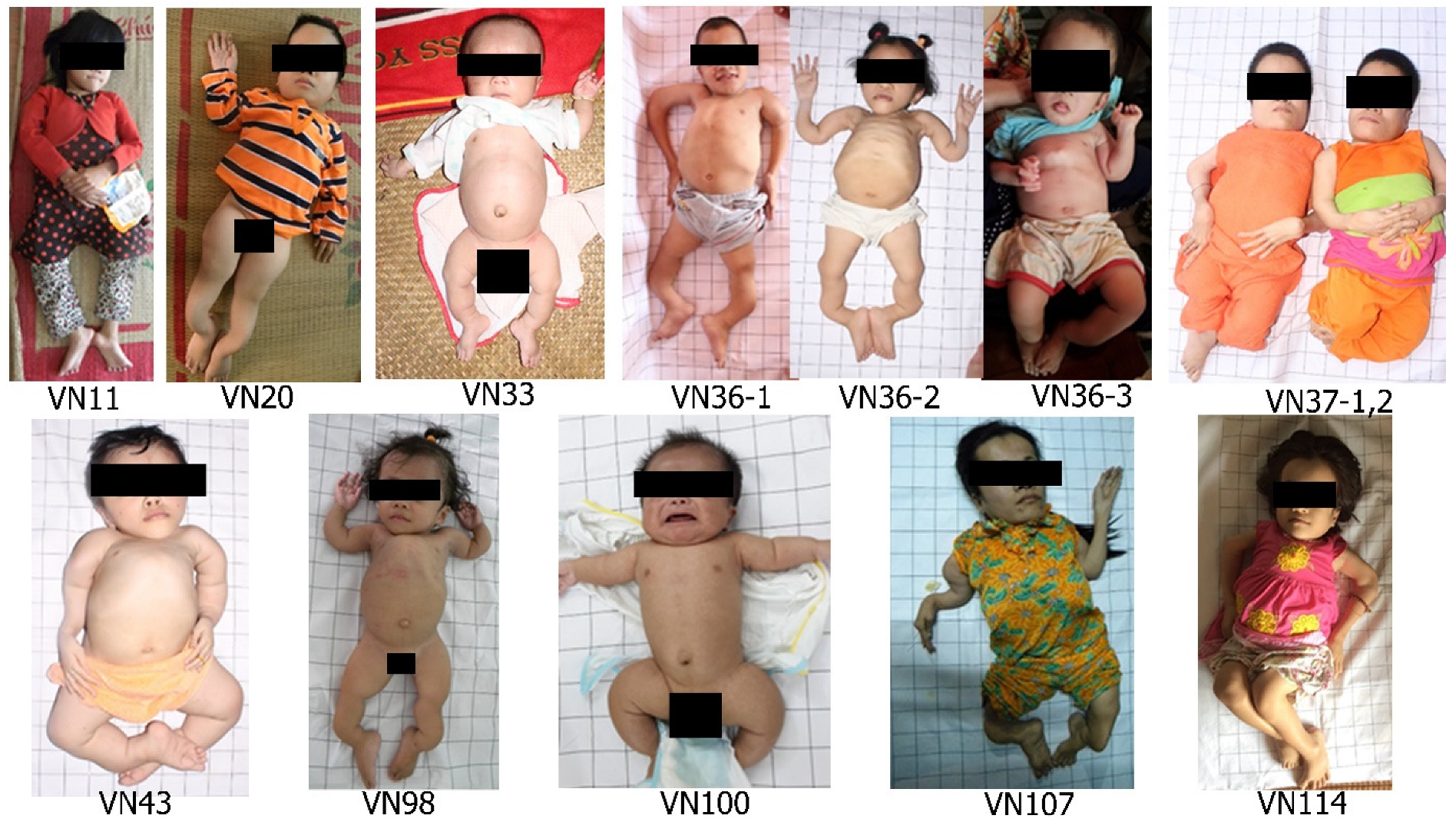
3.3. Clinical Characteristics
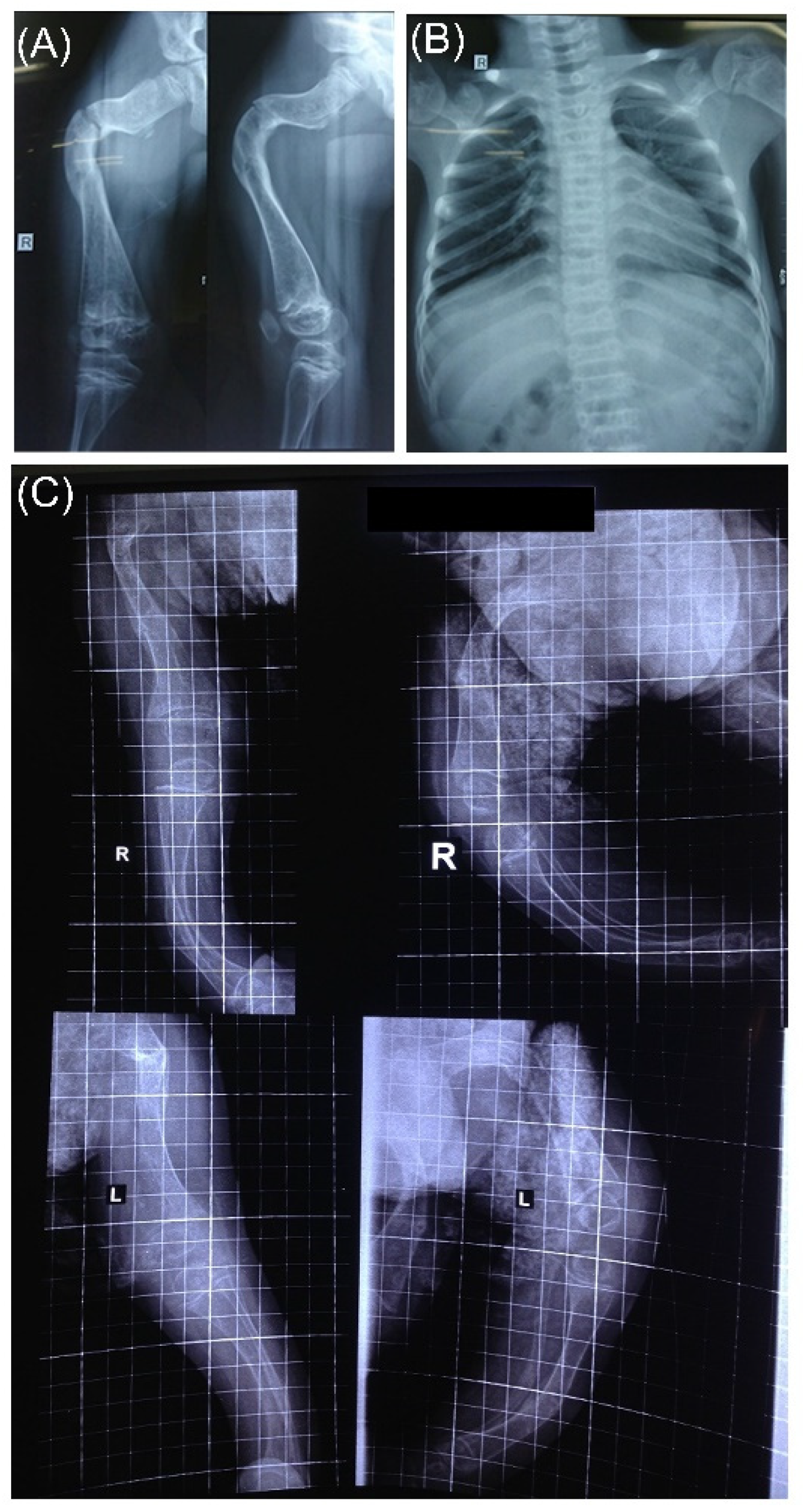
3.4. Radiographic Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Dijk, F.S.; Pals, G.; Van Rijn, R.R.; Nikkels, P.G.J.; Cobben, J.M. Classification of Osteogenesis Imperfecta revisited. Eur. J. Med. Genet. 2010, 53, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sillence, D.O.; Senn, A.; Danks, D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979, 16, 101–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMIM—Online Mendelian Inheritance in Man. Available online: https://omim.org/ (accessed on 23 February 2022).
- Claeys, L.; Storoni, S.; Eekhoff, M.; Elting, M.; Wisse, L.; Pals, G.; Bravenboer, N.; Maugeri, A.; Micha, D. Collagen transport and related pathways in Osteogenesis Imperfecta. Hum. Genet. 2021, 140, 1121–1141. [Google Scholar] [CrossRef] [PubMed]
- Essawi, O.; Symoens, S.; Fannana, M.; Darwish, M.; Farraj, M.; Willaert, A.; Essawi, T.; Callewaert, B.; De Paepe, A.; Malfait, F.; et al. Genetic analysis of osteogenesis imperfecta in the Palestinian population: Molecular screening of 49 affected families. Mol. Genet. Genom. Med. 2018, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Cabral, W.A.; Chang, W.; Barnes, A.M.; Weis, M.; Scott, M.A.; Leikin, S.; Makareeva, E.; Kuznetsova, N.V.; Rosenbaum, K.N.; Tifft, C.J.; et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007, 39, 359–365. [Google Scholar] [CrossRef]
- Santana, A.; Franzone, J.M.; McGreal, C.M.; Kruse, R.W.; Bober, M.B. A moderate form of osteogenesis imperfecta caused by compound heterozygous LEPRE1 mutations. Bone Rep. 2018, 9, 132–135. [Google Scholar] [CrossRef]
- Van Dijk, F.S.; Nesbitt, I.M.; Zwikstra, E.H.; Nikkels, P.G.J.; Piersma, S.R.; Fratantoni, S.A.; Jimenez, C.R.; Huizer, M.; Morsman, A.C.; Cobben, J.M.; et al. PPIB Mutations Cause Severe Osteogenesis Imperfecta. Am. J. Hum. Genet. 2009, 85, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP Is Required for Prolyl 3- Hydroxylation and Mutations Cause Recessive Osteogenesis Imperfecta. Cell 2006, 127, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Dalgleish, R. The human type I collagen mutation database. Nucleic Acids Res. 1997, 25, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Dejkhamron, P.; Intachai, W.; Ngamphiw, C.; Ketudat Cairns, J.R.; Kawasaki, K.; Ohazama, A.; Olsen, B.; Tongsima, S.; Angkurawaranon, S. A novel P3H1 mutation is associated with osteogenesis imperfecta type VIII and dental anomalies. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2021, 132, e198–e207. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cao, Y.; Wang, H.; Li, L.; Ren, X.; Mi, H.; Wang, Y.; Guan, Y.; Zhao, F.; Mao, B.; et al. Genotypic and Phenotypic Analysis in Chinese Cohort With Autosomal Recessive Osteogenesis Imperfecta. Front. Genet. 2020, 11, 984. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.M.; Bala, K.A. Severe cases of osteogenesis imperfecta type VIII due to a homozygous mutation in P3H1 (LEPRE1) and review of the literature. Adv. Clin. Exp. Med. 2021, 30. [Google Scholar] [CrossRef]
- Nadyrshina, D.; Zaripova, A.; Tyurin, A.; Minniakhmetov, I.; Zakharova, E.; Khusainova, R. Osteogenesis Imperfecta: Search for Mutations in Patients from the Republic of Bashkortostan (Russia). Genes 2022, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Tüysüz, B.; Elkanova, L.; Uludağ Alkaya, D.; Güleç, Ç.; Toksoy, G.; Güneş, N.; Yazan, H.; Bayhan, A.I.; Yıldırım, T.; Yeşil, G.; et al. Osteogenesis imperfecta in 140 Turkish families: Molecular spectrum and, comparison of long-term clinical outcome of those with COL1A1/A2 and biallelic variants. Bone 2022, 155, 116293. [Google Scholar] [CrossRef]
- Pepin, M.G.; Schwarze, U.; Singh, V.; Romana, M.; Jones-LeCointe, A.; Byers, P.H. Allelic background of LEPRE1 mutations that cause recessive forms of osteogenesis imperfecta in different populations. Mol. Genet. Genom. Med. 2013, 1, 194–205. [Google Scholar] [CrossRef]
- Cabral, W.A.; Barnes, A.M.; Adeyemo, A.; Cushing, K.; Chitayat, D.; Porter, F.D.; Panny, S.R.; Gulamali-Majid, F.; Tishkoff, S.A.; Rebbeck, T.R.; et al. A founder mutation in LEPRE1 carried by 1.5% of West Africans and 0.4% of African Americans causes lethal recessive osteogenesis imperfecta. Genet. Med. 2012, 14, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Baldridge, D.; Schwarze, U.; Morello, R.; Lennington, J.; Bertin, T.K.; Pace, J.M.; Pepin, M.G.; Weis, M.; Eyre, D.R.; Walsh, J.; et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008, 29, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Binh, H.D.; Maasalu, K.; Dung, V.C.; Ngoc, C.T.B.; Hung, T.T.; Nam, T.V.; Nhan, L.N.T.; Prans, E.; Reimann, E.; Zhytnik, L.; et al. The clinical features of osteogenesis imperfecta in Vietnam. Int. Orthop. 2017, 41, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Ho Duy, B.; Zhytnik, L.; Maasalu, K.; Kändla, I.; Prans, E.; Reimann, E.; Märtson, A.; Kõks, S. Mutation analysis of the COL1A1 and COL1A2 genes in Vietnamese patients with osteogenesis imperfecta. Hum. Genom. 2016, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhytnik, L.; Maasalu, K.; Duy, B.H.; Pashenko, A.; Khmyzov, S.; Reimann, E.; Prans, E.; Kõks, S.; Märtson, A. IFITM5 pathogenic variant causes osteogenesis imperfecta V with various phenotype severity in Ukrainian and Vietnamese patients. Hum. Genom. 2019, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2020; ISBN 9781491975190. [Google Scholar]
- Van Dijk, F.S.; Nikkels, P.G.J.; Den Hollander, N.S.; Nesbitt, I.M.; Van Rijn, R.R.; Cobben, J.M.; Pals, G. Lethal/severe osteogenesis imperfecta in a large family: A Novel Homozygous LEPRE1 Mutation and Bone Histological Findings. Pediatr. Dev. Pathol. 2011, 14, 228–234. [Google Scholar] [CrossRef]
- Willaert, A.; Malfait, F.; Symoens, S.; Gevaert, K.; Kayserili, H.; Megarbane, A.; Mortier, G.; Leroy, J.G.; Coucke, P.J.; De Paepe, A. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: Clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J. Med. Genet. 2009, 46, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moul, A.; Alladin, A.; Navarrete, C.; Abdenour, G.; Rodriguez, M.M. Osteogenesis Imperfecta Due to Compound Heterozygosity for the LEPRE1 Gene. Fetal Pediatr. Pathol. 2013, 32, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Ishii, T.; Barnes, A.M.; Weis, M.; Amano, N.; Tanaka, M.; Fukuzawa, R.; Nishimura, G.; Eyre, D.R.; Marini, J.C.; et al. A Novel Mutation in LEPRE1 That Eliminates Only the KDEL ER- Retrieval Sequence Causes Non-Lethal Osteogenesis Imperfecta. PLoS ONE 2012, 7, e36809. [Google Scholar] [CrossRef] [Green Version]
- Besio, R.; Garibaldi, N.; Leoni, L.; Cipolla, L.; Sabbioneda, S.; Biggiogera, M.; Mottes, M.; Aglan, M.; Otaify, G.A.; Temtamy, S.A.; et al. Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4-phenylbutyrate. DMM Dis. Model. Mech. 2019, 12, dmm038521. [Google Scholar] [CrossRef] [Green Version]
- Mrosk, J.; Gandham, S.B.; Shah, H.; Hecht, J.; Krüger, U.; Shukla, A.; Kornak, U.; Girisha, K.M. Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone 2018, 110, 368–377. [Google Scholar] [CrossRef]
- Zhang, Z.-L.; Zhang, H.; Ke, Y.; Yue, H.; Xiao, W.-J.; Yu, J.-B.; Gu, J.-M.; Hu, W.-W.; Wang, C.; He, J.-W.; et al. The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J. Bone Miner. Metab. 2012, 30, 69–77. [Google Scholar] [CrossRef]
- Skarp, S.; Xia, J.H.; Zhang, Q.; Löija, M.; Costantini, A.; Ruddock, L.W.; Mäkitie, O.; Wei, G.H.; Männikkö, M. Exome Sequencing Reveals a Phenotype Modifying Variant in ZNF528 in Primary Osteoporosis with a COL1A2 Deletion. J. Bone Miner. Res. 2020, 35, 2381–2392. [Google Scholar] [CrossRef]
- Pollitt, R.C.; Saraff, V.; Dalton, A.; Webb, E.A.; Shaw, N.J.; Sobey, G.J.; Mughal, M.Z.; Hobson, E.; Ali, F.; Bishop, N.J.; et al. Phenotypic variability in patients with osteogenesis imperfecta caused by BMP1 mutations. Am. J. Med. Genet. Part A 2016, 170, 3150–3156. [Google Scholar] [CrossRef] [Green Version]
- Zhytnik, L.; Maasalu, K.; Reimand, T.; Duy, B.H.; Kõks, S.; Märtson, A. Inter- and intrafamilial phenotypic variability in individuals with collagen-related Osteogenesis Imperfecta. Clin. Transl. Sci. 2020, 13, 960–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, M.; Campeau, P.M.; Lietman, C.D.; Lu, J.T.; Gibbs, R.A.; Schlesinger, A.E.; Lee, B.H. Osteogenesis imperfecta without features of type v caused by a mutation in the IFITM5 gene. J. Bone Miner. Res. 2013, 28, 2333–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, J.; Prockop, D.J. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu. Rev. Biophys. Biophys. Chem. 1991, 20, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Byers, P.H.; Wallis, G.A.; Willing, M.C. Osteogenesis imperfecta: Translation of mutation to phenotype. J. Med. Genet. 1991, 28, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Patient ID | Age (y.o.) | Sex | Type | Total Fractures | Fractures per Year | Intrauterine Fractures | Chest Deformities | Brachycephaly | Blue Sclera | Hearing Loss | DI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | VN10 | 13 | F | 1 | 0 | 0 | No | No | No | Yes | No | Yes |
| 2 | VN11 | 10 | F | 4 | 27 | 2.7 | No | Yes | Yes | Yes | Yes | Yes |
| 3 | VN20 | 10 | M | 3 | 10 | 1.0 | No | Yes | Yes | Yes | No | Yes |
| 4 | VN33 | 1 | M | 4 | 8 | 8.0 | No | No | Yes | Yes | No | No |
| 5 | VN36-1 | 16 | M | 3 | 15 | 0.9 | Yes | Yes | Yes | Yes | No | No |
| 6 | VN36-2 | 12 | F | 3 | 14 | 1.2 | Yes | Yes | Yes | Yes | No | Yes |
| 7 | VN36-3 | 4 | M | 4 | 6 | 1.5 | Yes | Yes | Yes | Yes | No | No |
| 8 | VN37-1 | 35 | F | 3 | 20 | 0.6 | Yes | Yes | Yes | Yes | No | No |
| 9 | VN37-2 | 31 | F | 3 | 18 | 0.6 | Yes | Yes | Yes | Yes | No | No |
| 10 | VN42 | 11 | F | 4 | 25 | 2.3 | No | No | No | Yes | No | No |
| 11 | VN43 | 12 | F | 3 | 40 | 3.3 | Yes | Yes | Yes | Yes | No | Yes |
| 12 | VN75 | 5 | M | 3 | 14 | 2.8 | No | Yes | Yes | Yes | No | Yes |
| 13 | VN98 | 5 | F | 3 | 20 | 4.0 | Yes | Yes | Yes | No | No | No |
| 14 | VN100 | 0.25 | M | 4 | 2 | NA | Yes | No | No | Yes | No | No |
| 15 | VN101 | 22 | F | 3 | 40 | 1.8 | Yes | Yes | Yes | No | No | Yes |
| 16 | VN107 | 33 | F | 3 | 30 | 0.9 | Yes | Yes | Yes | No | Yes | No |
| 17 | VN114 | 12 | F | 3 | 15 | 1.3 | No | Yes | Yes | Yes | No | Yes |
| № | Patient ID | Variant | Variant State | Variant Type | Protein Effect | ACMG Classification | Variant in the Mother | Variant in the Father |
|---|---|---|---|---|---|---|---|---|
| 1 | VN10 | c.1170+5G>C c.2197A>G# | CH * | Splice siteMissense | p.? p.(Lys733Glu) | P—PS3, PP3, PM2, PS4 LP—PM2, PM6, PM1, (PM3) | c.1170+5G>C heterozygous | - |
| 2 | VN11 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 3 | VN20 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 4 | VN33 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 5 | VN36-1 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 6 | VN36-2 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 7 | VN36-3 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 8 | VN37-1 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 9 | VN37-2 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 10 | VN42 | c.1170+5G>C c.1934G>A# | CH | Splice site Missense | p.? p.(Cys645Tyr) | P—PS3, PP3, PM2, PS4 LP—PM3, PM2, PP3 | c.1934G>A heterozygous | c.1170+5G>C heterozygous |
| 11 | VN43 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 12 | VN75 | c.1224-79G>A# c.257del# | CH | Intronic Frameshift | p.(?) p.(Glu86Glyfs*22) | LP—PM3, PM2, PP3, (PS4) P—PM3, PVS1, PM2 | c.257del heterozygous | c.1224-79G>A heterozygous |
| 13 | VN98 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 14 | VN100 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
| 15 | VN101 | c.1224-79G>A# | H | Intronic | p.(?) | LP—PM3, PM2, PP3, (PS4) | NA | NA |
| 16 | VN107 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | heterozygous | NA |
| 17 | VN114 | c.1170+5G>C | H | Splice site | p.? | P—PS3, PP3, PM2, PS4 | c.1170+5G>C heterozygous | c.1170+5G>C heterozygous |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhytnik, L.; Duy, B.H.; Eekhoff, M.; Wisse, L.; Pals, G.; Reimann, E.; Kõks, S.; Märtson, A.; Maugeri, A.; Maasalu, K.; et al. Phenotypic Variation in Vietnamese Osteogenesis Imperfecta Patients Sharing a Recessive P3H1 Pathogenic Variant. Genes 2022, 13, 407. https://doi.org/10.3390/genes13030407
Zhytnik L, Duy BH, Eekhoff M, Wisse L, Pals G, Reimann E, Kõks S, Märtson A, Maugeri A, Maasalu K, et al. Phenotypic Variation in Vietnamese Osteogenesis Imperfecta Patients Sharing a Recessive P3H1 Pathogenic Variant. Genes. 2022; 13(3):407. https://doi.org/10.3390/genes13030407
Chicago/Turabian StyleZhytnik, Lidiia, Binh Ho Duy, Marelise Eekhoff, Lisanne Wisse, Gerard Pals, Ene Reimann, Sulev Kõks, Aare Märtson, Alessandra Maugeri, Katre Maasalu, and et al. 2022. "Phenotypic Variation in Vietnamese Osteogenesis Imperfecta Patients Sharing a Recessive P3H1 Pathogenic Variant" Genes 13, no. 3: 407. https://doi.org/10.3390/genes13030407
APA StyleZhytnik, L., Duy, B. H., Eekhoff, M., Wisse, L., Pals, G., Reimann, E., Kõks, S., Märtson, A., Maugeri, A., Maasalu, K., & Micha, D. (2022). Phenotypic Variation in Vietnamese Osteogenesis Imperfecta Patients Sharing a Recessive P3H1 Pathogenic Variant. Genes, 13(3), 407. https://doi.org/10.3390/genes13030407