
HNF1A POU Domain Mutations Found in Japanese Liver Cancer Patients Cause Downregulation of HNF4A Promoter Activity with Possible Disruption in Transcription Networks

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Primers
2.3. Reporter Assay
2.4. Western Blotting
2.5. Immunofluorescence (IFC)
2.6. Electrophoretic Mobility-Shift Assay (EMSA)
2.7. Molecular Dynamics (MD) Simulations
2.8. KD by HNF4A siRNA
2.9. RNA-Sequencing (RNA-Seq) and Functional Analysis
2.10. Statistical Analyses
3. Results
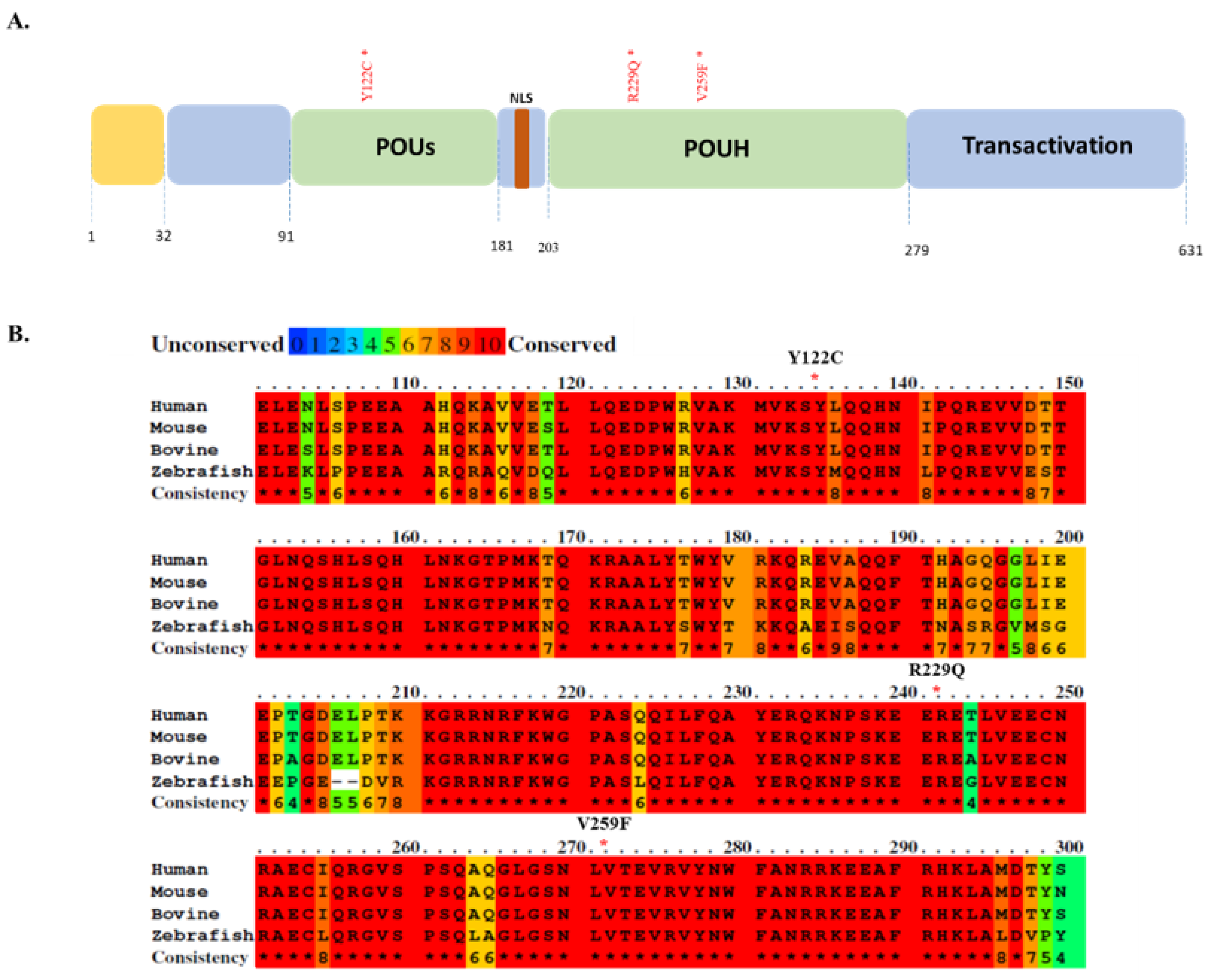
3.1. Somatic Mutations Found in the Functional Domain of HNF1A
3.2. HNF1A Mutants Display Reduced Transcriptional Activity and Decreased Binding Ability
3.3. Dynamics of the HNF1A V259F Mutant Revealed That the Mutation Affects Protein Stability and Causes Rearrangement in the N-Terminal Region
3.4. siRNA KD of HNF4A Causes Differential Gene Expression and Overrepresented Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DEGs | Differentially expressed genes |
| DMEM | Dulbecco’s modified Eagle’s medium |
| EMSA | Electrophoretic mobility-shift assay |
| FBS | Fetal bovine serum |
| GO | Gene ontology |
| HCC | Hepatocellular carcinoma |
| HNF1A | Hepatocyte nuclear factor 1A |
| HNF4A | Hepatocyte nuclear factor 4A |
| HEK293 | Human embryonic kidney cells 293 |
| Huh7 | Human hepatoma cell line |
| IFC | Immunofluorescence |
| ICGC | International Cancer Genome Consortium |
| KD | Knockdown |
| KO | Knockout |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MODY3 | Maturity-onset diabetes of the young type 3 |
| MD | Molecular dynamics |
| NGS | Next-generation sequencing |
| NASH | Non-alcoholic steatohepatitis |
| Hepa1-6 | Non-immunogenic mouse hepatoma cells |
| NF-κB | Nuclear factor kappa-B |
| POUh | Pit-Oct-Unc (POU) homeodomain |
| POUs | Pit-Oct-Unc (POU) specific domain |
| RMSF | Root-mean-square fluctuation |
| RMSD | Root-mean-square deviation |
| TCGA | The Cancer Genome Atlas |
| LINCS | The Library of Integrated Network-Based Cellular Signatures |
| VMD | Visual Molecular Dynamics |
| WB | Western blot |
| Wnt | Wingless-related integration site |
References
- Lin, L.; Yan, L.; Liu, Y.; Qu, C.; Ni, J.; Li, H. The Burden and Trends of Primary Liver Cancer Caused by Specific Etiologies from 1990 to 2017 at the Global, Regional, National, Age, and Sex Level Results from the Global Burden of Disease Study 2017. Liver Cancer 2020, 9, 563–582. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational Signatures Associated with Tobacco Smoking in Human Cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-Genome Mutational Landscape and Characterization of Noncoding and Structural Mutations in Liver Cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- D’Angelo, A.; Bluteau, O.; Garcia-Gonzalez, M.A.; Gresh, L.; Doyen, A.; Garbay, S.; Robine, S.; Pontoglio, M. Hepatocyte nuclear factor 1alpha and beta control terminal differentiation and cell fate commitment in the gut epithelium. Development 2010, 137, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Blumenfeld, M.; Maury, M.; Chouard, T.; Yaniv, M.; Condamine, H. Hepatic Nuclear Factor 1 (HNF1) shows a Wider Distribution than Products of its Known Target Genes in Developing Mouse. Development 1991, 113, 589–599. [Google Scholar] [CrossRef]
- Lau, H.H.; Ng, N.H.J.; Loo, L.S.W.; Jasmen, J.B.; Teo, A.K.K. The Molecular Functions of Hepatocyte Nuclear Factors—in and Beyond the Liver. J. Hepatol. 2018, 68, 1033–1048. [Google Scholar] [CrossRef] [Green Version]
- Ni, Q.; Ding, K.; Wang, K.Q.; He, J.; Yin, C.; Shi, J.; Zhang, X.; Xie, W.F.; Shi, Y.Q. Deletion of HNF1α in hepatocytes results in fatty liver-related hepatocellular carcinoma in mice. FEBS Lett. 2017, 591, 1947–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krützfeldt, J.; Rösch, N.; Hausser, J.; Manoharan, M.; Zavolan, M.; Stoffel, M. MicroRNA-194 is a target of transcription factor 1 (Tcf1, HNF1α) in adult liver and controls expression of frizzled-6. Hepatology 2012, 55, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, C.; Li, Y.; Huan, L.; Zhang, Y.; Zhao, F.; Wang, Q.; Liang, L.; Ding, J.; Liu, L.; Chen, T.; et al. NF-κB signaling relieves negative regulation by miR-194 in hepatocellular carcinoma by suppressing the transcription factor HNF-1α. Sci. Signal. 2015, 8, 75. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, Y.; Yin, C.; Zhang, X.; Ning, B.F.; Zhang, Q.; Zhang, J.P.; Qiu, L.; Qin, X.R.; Chen, Y.X.; et al. Recombinant adenovirus carrying the hepatocyte nuclear factor-1alpha gene inhibits hepatocellular carcinoma xenograft growth in mice. Hepatology 2011, 54, 2036–2047. [Google Scholar] [CrossRef] [PubMed]
- Fujino, S.; Miyoshi, N.; Ito, A.; Yasui, M.; Matsuda, C.; Ohue, M.; Uemura, M.; Mizushima, T.; Doki, Y.; Eguchi, H. HNF1A Regulates Colorectal Cancer Progression and Drug Resistance as a Downstream of POU5F1. Sci. Rep. 2021, 11, 10363. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Liu, L.; Yang, W.; Zhang, Q. HNF1A-AS1 Regulates Cell Migration, Invasion and Glycolysis via Modulating miR-124/MYO6 in Colorectal Cancer Cells. OncoTargets Ther. 2020, 13, 1507–1518. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Li, Y.; Wang, H.; Fleming, J.; Li, M.; Kang, Y.; Zhang, R.; Li, D. Hepatocyte Nuclear Factor 1A (HNF1A) as a Possible Tumor Suppressor in Pancreatic Cancer. PLoS ONE 2015, 10, e0121082. [Google Scholar]
- Wang, X.; Hassan, W.; Zhao, J.; Bakht, S.; Nie, Y.; Wang, Y.; Pang, Q.; Huang, Z. The Impact of Hepatocyte Nuclear Factor-1alpha on Liver Malignancies and Cell Stemness with Metabolic Consequences. Stem Cell. Res. Ther. 2019, 10, 315. [Google Scholar] [CrossRef] [Green Version]
- Cereghini, S. Liver-Enriched Transcription Factors and Hepatocyte Differentiation. FASEB J. 1996, 10, 267–282. [Google Scholar] [CrossRef]
- Mendel, D.B.; Hansen, L.P.; Graves, M.K.; Conley, P.B.; Crabtree, G.R. HNF-1 Alpha and HNF-1 Beta (vHNF-1) Share Dimerization and Homeo Domains, but Not Activation Domains, and Form Heterodimers in Vitro. Genes Dev. 1991, 5, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Moller, A.M.; Dalgaard, L.T.; Pociot, F.; Nerup, J.; Hansen, T.; Pedersen, O. Mutations in the Hepatocyte Nuclear Factor-1alpha Gene in Caucasian Families Originally Classified as having Type I Diabetes. Diabetologia 1998, 41, 1528–1531. [Google Scholar] [PubMed] [Green Version]
- Hansen, S.K.; Parrizas, M.; Jensen, M.L.; Pruhova, S.; Ek, J.; Boj, S.F.; Johansen, A.; Maestro, M.A.; Rivera, F.; Eiberg, H.; et al. Genetic Evidence that HNF-1alpha-Dependent Transcriptional Control of HNF-4alpha is Essential for Human Pancreatic Beta Cell Function. J. Clin. Investig. 2002, 110, 827–833. [Google Scholar] [CrossRef]
- Piccolo, P.; Annunziata, P.; Soria, L.R.; Attanasio, S.; Barbato, A.; Castello, R.; Carissimo, A.; Quagliata, L.; Terracciano, L.M.; Brunetti-Pierri, N. Down-Regulation of Hepatocyte Nuclear Factor-4alpha and Defective Zonation in Livers Expressing Mutant Z alpha1-Antitrypsin. Hepatology 2017, 66, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.J.; Mendel, D.B.; Hansen, L.P.; Crabtree, G.R. Independent Regulation of HNF-1 Alpha and HNF-1 Beta by Retinoic Acid in F9 Teratocarcinoma Cells. EMBO J. 1991, 10, 2231–2236. [Google Scholar] [CrossRef] [PubMed]
- Kyithar, M.P.; Bonner, C.; Bacon, S.; Kilbride, S.M.; Schmid, J.; Graf, R.; Prehn, J.H.; Byrne, M.M. Effects of Hepatocyte Nuclear Factor-1A and -4A on Pancreatic Stone protein/regenerating Protein and C-Reactive Protein Gene Expression: Implications for Maturity-Onset Diabetes of the Young. J. Transl. Med. 2013, 11, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, H.; Fujimoto, A.; Kono, H.; Furuta, M.; Fujita, M.; Nakagawa, H. Loss-of-Function Mutations in Zn-Finger DNA-Binding Domain of HNF4A Cause Aberrant Transcriptional Regulation in Liver Cancer. Oncotarget 2018, 9, 26144–26156. [Google Scholar] [CrossRef] [Green Version]
- Eeckhoute, J.; Formstecher, P.; Laine, B. Hepatocyte Nuclear Factor 4alpha Enhances the Hepatocyte Nuclear Factor 1alpha-Mediated Activation of Transcription. Nucleic Acids Res. 2004, 32, 2586–2593. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H.; Jaschkowitz, K.; Bulman, M.; Frayling, T.M.; Mitchell, S.M.; Roosen, S.; Lingott-Frieg, A.; Tack, C.J.; Ellard, S.; Ryffel, G.U.; et al. A Distant Upstream Promoter of the HNF-4alpha Gene Connects the Transcription Factors Involved in Maturity-Onset Diabetes of the Young. Hum. Mol. Genet. 2001, 10, 2089–2097. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Okada, Y.; Boroevich, K.A.; Tsunoda, T.; Taniguchi, H.; Nakagawa, H. Systematic Analysis of Mutation Distribution in Three Dimensional Protein Structures Identifies Cancer Driver Genes. Sci. Rep. 2016, 6, 26483. [Google Scholar] [CrossRef] [Green Version]
- Hechtman, J.F.; Abou-Alfa, G.K.; Stadler, Z.K.; Mandelker, D.L.; Roehrl, M.H.A.; Zehir, A.; Vakiani, E.; Middha, S.; Klimstra, D.S.; Shia, J. Somatic HNF1A Mutations in the Malignant Transformation of Hepatocellular Adenomas: A Retrospective Analysis of Data from MSK-IMPACT and TCGA. Hum. Pathol. 2019, 83, 1–6. [Google Scholar] [CrossRef]
- Bjorkhaug, L.; Ye, H.; Horikawa, Y.; Sovik, O.; Molven, A.; Njolstad, P.R. MODY Associated with Two Novel Hepatocyte Nuclear Factor-1alpha Loss-of-Function Mutations (P112L and Q466X). Biochem. Biophys. Res. Commun. 2000, 279, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.H.; Deng, L.F.; Chen, F.; Ding, K.; Chen, W.S.; Xie, W.F.; Zhang, X.P. Q511L Mutation of HNF1alpha in Hepatocellular Carcinoma Suppresses the Transcriptional Activity and the Anti-Tumor Effect of HNF1alpha. Biochem. Biophys. Res. Commun. 2018, 495, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, O.; Jeannot, E.; Bioulac-Sage, P.; Marqués, J.M.; Blanc, J.F.; Bui, H.; Beaudoin, J.C.; Franco, D.; Balabaud, C.; Laurent-Puig, P.; et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat. Genet. 2002, 32, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.I.; Frantz, J.D.; Oh, B.C.; Hansen, L.; Dhe-Paganon, S.; Shoelson, S.E. Diabetes Mutations Delineate an Atypical POU Domain in HNF-1alpha. Mol. Cell 2002, 10, 1129–1137. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, I.S.; Cheatham, T.E., 3rd. Determination of Alkali and Halide Monovalent Ion Parameters for use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B. 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; de Vries, A.H.; Marrink, S.J.; Berendsen, H.J. Efficient Algorithms for Langevin and DPD Dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLR algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bjorkhaug, L.; Sagen, J.V.; Thorsby, P.; Sovik, O.; Molven, A.; Njolstad, P.R. Hepatocyte Nuclear Factor-1 Alpha Gene Mutations and Diabetes in Norway. J. Clin. Endocrinol. Metab. 2003, 88, 920–931. [Google Scholar] [CrossRef]
- Galan, M.; Garcia-Herrero, C.M.; Azriel, S.; Gargallo, M.; Duran, M.; Gorgojo, J.J.; Andia, V.M.; Navas, M.A. Differential Effects of HNF-1alpha Mutations Associated with Familial Young-Onset Diabetes on Target Gene Regulation. Mol. Med. 2011, 17, 256–265. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address: [email protected], & Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar]
- Plengvidhya, N.; Tangjittipokin, W.; Teerawattanapong, N.; Narkdontri, T.; Yenchitsomanus, P.T. HNF1A mutation in a Thai patient with maturity-onset diabetes of the young: A case report. World J. Diabetes 2019, 10, 414–420. [Google Scholar] [CrossRef]
- Beysel, S.; Eyerci, N.; Pinarli, F.A.; Kizilgul, M.; Ozcelik, O.; Caliskan, M.; Cakal, E. HNF1A gene p.I27L is associated with early-onset, maturity-onset diabetes of the young-like diabetes in Turkey. BMC Endocr. Disord. 2019, 19, 51. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K.; Bjørkhaug, L.; Mahajan, S.; Kanthimathi, S.; Njølstad, P.R.; Srinivasan, N.; Mohan, V.; Radha, V. Structure-function studies of HNF1A (MODY3) gene mutations in South Indian patients with monogenic diabetes. Clin. Genet. 2016, 90, 486–495. [Google Scholar] [CrossRef] [PubMed]
- P, S.; D, T.K.; C, G.P.D.; R, S.; Zayed, H. Determining the Role of Missense Mutations in the POU Domain of HNF1A that Reduce the DNA-Binding Affinity: A Computational Approach. PLoS ONE 2017, 12, e0174953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.; Song, L.; Yang, Y.; Tian, L.; Li, X.; Wu, S.; Huang, W.; Ren, H.; Tang, N.; Ding, K. Validation of a multi-omics strategy for prioritizing personalized candidate driver genes. Oncotarget 2016, 7, 38440–38450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najmi, L.A.; Aukrust, I.; Flannick, J.; Molnes, J.; Burtt, N.; Molven, A.; Groop, L.; Altshuler, D.; Johansson, S.; Bjorkhaug, L.; et al. Functional Investigations of HNF1A Identify Rare Variants as Risk Factors for Type 2 Diabetes in the General Population. Diabetes 2017, 66, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.H.; Lu, S.X.; Liu, L.L.; Zhang, C.Z.; Yun, J.P. Increased Expression of Hepatocyte Nuclear Factor 4 Alpha Transcribed by Promoter 2 Indicates a Poor Prognosis in Hepatocellular Carcinoma. Ther. Adv. Gastroenterol. 2017, 10, 761–771. [Google Scholar] [CrossRef]
- Servitja, J.M.; Pignatelli, M.; Maestro, M.A.; Cardalda, C.; Boj, S.F.; Lozano, J.; Blanco, E.; Lafuente, A.; McCarthy, M.I.; Sumoy, L.; et al. Hnf1alpha (MODY3) Controls Tissue-Specific Transcriptional Programs and Exerts Opposed Effects on Cell Growth in Pancreatic Islets and Liver. Mol. Cell. Biol. 2009, 29, 2945–2959. [Google Scholar] [CrossRef] [Green Version]
- Walesky, C.; Edwards, G.; Borude, P.; Gunewardena, S.; O’Neil, M.; Yoo, B.; Apte, U. Hepatocyte Nuclear Factor 4 Alpha Deletion Promotes Diethylnitrosamine-Induced Hepatocellular Carcinoma in Rodents. Hepatology 2013, 57, 2480–2490. [Google Scholar] [CrossRef] [Green Version]
- Harries, L.W.; Brown, J.E.; Gloyn, A.L. Species-Specific Differences in the Expression of the HNF1A, HNF1B and HNF4A Genes. PLoS ONE 2009, 4, e7855. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, A.; Kannari, M.; Tsuchida, Y.; Sasaki, S.; Saito, C.; Matsuta, T.; Maeda, T.; Akiyama, M.; Nakamura, T.; Sakaguchi, M.; et al. An HNF4alpha-microRNA-194/192 Signaling Axis Maintains Hepatic Cell Function. J. Biol. Chem. 2017, 292, 10574–10585. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, L.; Marchetti, A.; Cicchini, C.; Conigliaro, A.; Conti, B.; Mancone, C.; Bonzo, J.A.; Gonzalez, F.J.; Alonzi, T.; Amicone, L.; et al. The Stable Repression of Mesenchymal Program is Required for Hepatocyte Identity: A Novel Role for Hepatocyte Nuclear Factor 4alpha. Hepatology 2011, 53, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Strub, C.; Alies, C.; Lougarre, A.; Ladurantie, C.; Czaplicki, J.; Fournier, D. Mutation of Exposed Hydrophobic Amino Acids to Arginine to Increase Protein Stability. BMC Biochem. 2004, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Cai, X.; Cheng, S.; Zhou, H.; Zhang, Z.; Ren, J.; Ren, F.; Yang, Q.; Tao, N.; Chen, J. Ornithine transcarbamylase downregulation is associated with poor prognosis in hepatocellular carcinoma. Oncol. Lett. 2019, 17, 5030–5038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, D.; Jin, H.; Jin, G.; Wang, C.; Wang, N.; Hu, F.; Luo, Q.; Chu, W.; Yao, M.; Qin, W. The asialoglycoprotein receptor suppresses the metastasis of hepatocellular carcinoma via LASS2-mediated inhibition of V-ATPase activity. Cancer Lett. 2016, 379, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Shchelochkov, O.A.; Gallagher, R.C.; Batshaw, M.L. Hepatocellular carcinoma in a research subject with ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2012, 105, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Bell, P.; Morizono, H.; He, Z.; Pumbo, E.; Yu, H.; White, J.; Batshaw, M.L.; Wilson, J.M. AAV gene therapy corrects OTC deficiency and prevents liver fibrosis in aged OTC-knock out heterozygous mice. Mol. Genet. Metab. 2017, 120, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.T.; Henderson, C.A.; Warren, C.R.; Friesen, M.; Xia, F.; Becker, C.E.; Musunuru, K.; Cowan, C.A. Asialoglycoprotein receptor 1 is a specific cell-surface marker for isolating hepatocytes derived from human pluripotent stem cells. Development 2016, 143, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Fang, S.; Weng, Q.; Lv, X.; Meng, M.; Zhu, J.; Zheng, L.; Hu, Y.; Gao, Y.; Wu, X.; et al. Integrated analysis reveals critical glycolytic regulators in hepatocellular carcinoma. Cell Commun. Signal. 2020, 18, 97. [Google Scholar] [CrossRef]
- Kuhajda, F.P.; Piantadosi, S.; Pasternack, G.R. Haptoglobin-related protein (Hpr) epitopes in breast cancer as a predictor of recurrence of the disease. N. Engl. J. Med. 1989, 321, 636–641. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, G.; Dong, B.; Piao, J.; Chen, L.; Lin, Z. The NQO1/PKLR axis promotes lymph node metastasis and breast cancer progression by modulating glycolytic reprogramming. Cancer lett. 2019, 453, 170–183. [Google Scholar] [CrossRef]
- Fang, L.; Che, Y.; Zhang, C.; Huang, J.; Lei, Y.; Lu, Z.; Sun, N.; He, J. PLAU directs conversion of fibroblasts to inflammatory cancer-associated fibroblasts, promoting esophageal squamous cell carcinoma progression via uPAR/Akt/NF-κB/IL8 pathway. Cell Death Discov. 2021, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Staniczek, T.; Kurschner, S.W.; Schmid, C.D.; Schonhaber, H.; Cordero, J.; Kessler, L.; Mathes, A.; Sticht, C.; Nessling, M.; et al. Endothelial GATA4 Controls Liver Fibrosis and Regeneration by Preventing a Pathogenic Switch in Angiocrine Signaling. J. Hepatol. 2021, 74, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Marinho, A.T.; Lu, H.; Pereira, S.A.; Monteiro, E.; Gabra, H.; Recchi, C. Anti-Tumorigenic and Platinum-Sensitizing Effects of Apolipoprotein A1 and Apolipoprotein A1 Mimetic Peptides in Ovarian Cancer. Front. Pharmacol. 2019, 9, 1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ma, Z.; Jiang, S.; Hu, W.; Li, T.; Di, S.; Wang, D.; Yang, Y. A Global Perspective on FOXO1 in Lipid Metabolism and Lipid-Related Diseases. Prog. Lipid Res. 2017, 66, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Enane, F.O.; Shuen, W.H.; Gu, X.; Quteba, E.; Przychodzen, B.; Makishima, H.; Bodo, J.; Ng, J.; Chee, C.L.; Ba, R.; et al. GATA4 loss of function in liver cancer impedes precursor to hepatocyte transition. J. Clin. Investig. 2017, 127, 3527–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Ma, H.; Ge, X.; Edwards, P.A.; Zhang, Y. Hepatic hepatocyte nuclear factor 4α is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 328–336. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation ID | Genomic DNA Change | Type | Consequences | Project in Which Mutation Observed | Conservation among Species |
|---|---|---|---|---|---|
| MU854410 | chr12:g.121432028G>T | Single base substitution | V259F | LINC-JP | YES |
| MU837628 | chr12:g.121426674A>G | Single base substitution | Y122C | LINC-JP | YES |
| MU81565444 | chr12:g.121431445G>T | Single base substitution | A217S | LICA-CN | YES |
| MU85877851 | chr12:g.121426663G>T | Single base substitution | M118I | LICA-CN | YES |
| MU20638 | chr12:g.121431482G>A | Single base substitution | R229Q | LICA-FR | YES |
| MU29769426 | chr12:g.121431410A>C | Single base substitution | K205T | LICA-FR | YES |
| MU82396333 | chr12:g.121426664G>T | Single base substitution | V119F | LICA-CN | YES |
| MU602436 | chr12:g.121426701G>T | Single base substitution | R131L | LICA-CN | YES |
| MU29793014 | chr12:g.121431983A>G | Single base substitution | R244G | LIHC-US | YES |
| MU29769474 | chr12:g.121431977A>T | Single base substitution | I242F | LICA-FR | Not in Zebrafish |
| MU128970370 | chr12:g.121426782A>G | Single base substitution | K158R | LIHC-US | YES |
| MU85320917 | chr12:g.121431501G>T | Single base substitution | E235D | LICA-CN | YES |
| MU128971993 | chr12:g.121431424T>A | Single base substitution | S210T | LIHC-US | YES |
| MU29496420 | chr12:g.121432040C>G | Single base substitution | R263G | LIHC-US | YES |
| MU29433874 | chr12:g.121432014T>A | Single base substitution | L254Q | LIHC-US | YES |
| MU29746856 | chr12:g.121431506A>G | Single base substitution | N237S | LIHC-US | YES |
| MU822656 | chr12:g.121431466C>G | Single base substitution | P224A | LIAD-FR | YES |
| MU822434 | chr12:g.121431413G>T | Single base substitution | W206L | LIAD-FR | YES |
| MU822864 | chr12:g.121432067C>A | Single base substitution | R272S | LIAD-FR | YES |
| MU823044 | chr12:g.121432041G>T | Single base substitution | R263L | LIAD-FR | YES |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, E.; Teeli, A.S.; Winiarczyk, D.; Taguchi, M.; Sakuraba, S.; Kono, H.; Leszczyński, P.; Pierzchała, M.; Taniguchi, H. HNF1A POU Domain Mutations Found in Japanese Liver Cancer Patients Cause Downregulation of HNF4A Promoter Activity with Possible Disruption in Transcription Networks. Genes 2022, 13, 413. https://doi.org/10.3390/genes13030413
Haque E, Teeli AS, Winiarczyk D, Taguchi M, Sakuraba S, Kono H, Leszczyński P, Pierzchała M, Taniguchi H. HNF1A POU Domain Mutations Found in Japanese Liver Cancer Patients Cause Downregulation of HNF4A Promoter Activity with Possible Disruption in Transcription Networks. Genes. 2022; 13(3):413. https://doi.org/10.3390/genes13030413
Chicago/Turabian StyleHaque, Effi, Aamir Salam Teeli, Dawid Winiarczyk, Masahiko Taguchi, Shun Sakuraba, Hidetoshi Kono, Paweł Leszczyński, Mariusz Pierzchała, and Hiroaki Taniguchi. 2022. "HNF1A POU Domain Mutations Found in Japanese Liver Cancer Patients Cause Downregulation of HNF4A Promoter Activity with Possible Disruption in Transcription Networks" Genes 13, no. 3: 413. https://doi.org/10.3390/genes13030413