
Tumor Heterogeneity and Molecular Characteristics of Glioblastoma Revealed by Single-Cell RNA-Seq Data Analysis



Abstract
:1. Introduction
2. Method
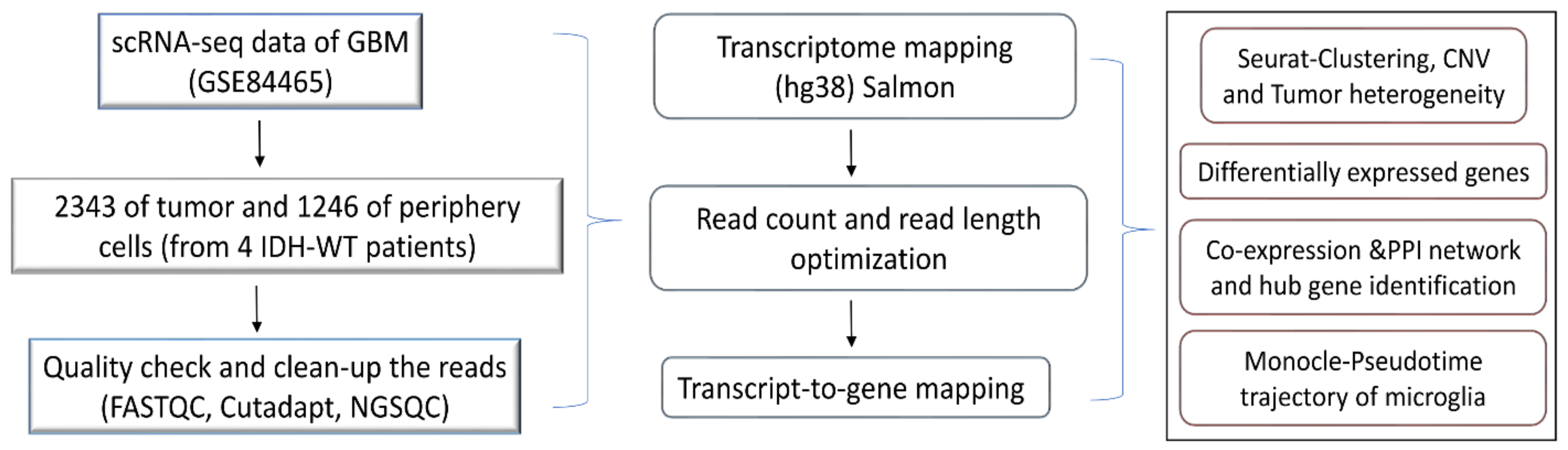
2.1. Data Collection and Quality Check
2.2. Dataset Preprocessing by Seurat
2.3. Dimensionality Reduction and Cluster Identification
2.4. Determination of Copy Number Variations (CNVs)
2.5. Differential Gene Expression and Functional Annotation
2.6. Network Analysis
2.7. Monocle Pseudotime Trajectory Reconstruction and Analysis
3. Results
3.1. Cell Clusters and Tumor Heterogeneity in GBM
3.1.1. Cell Type Identifications of the Clusters
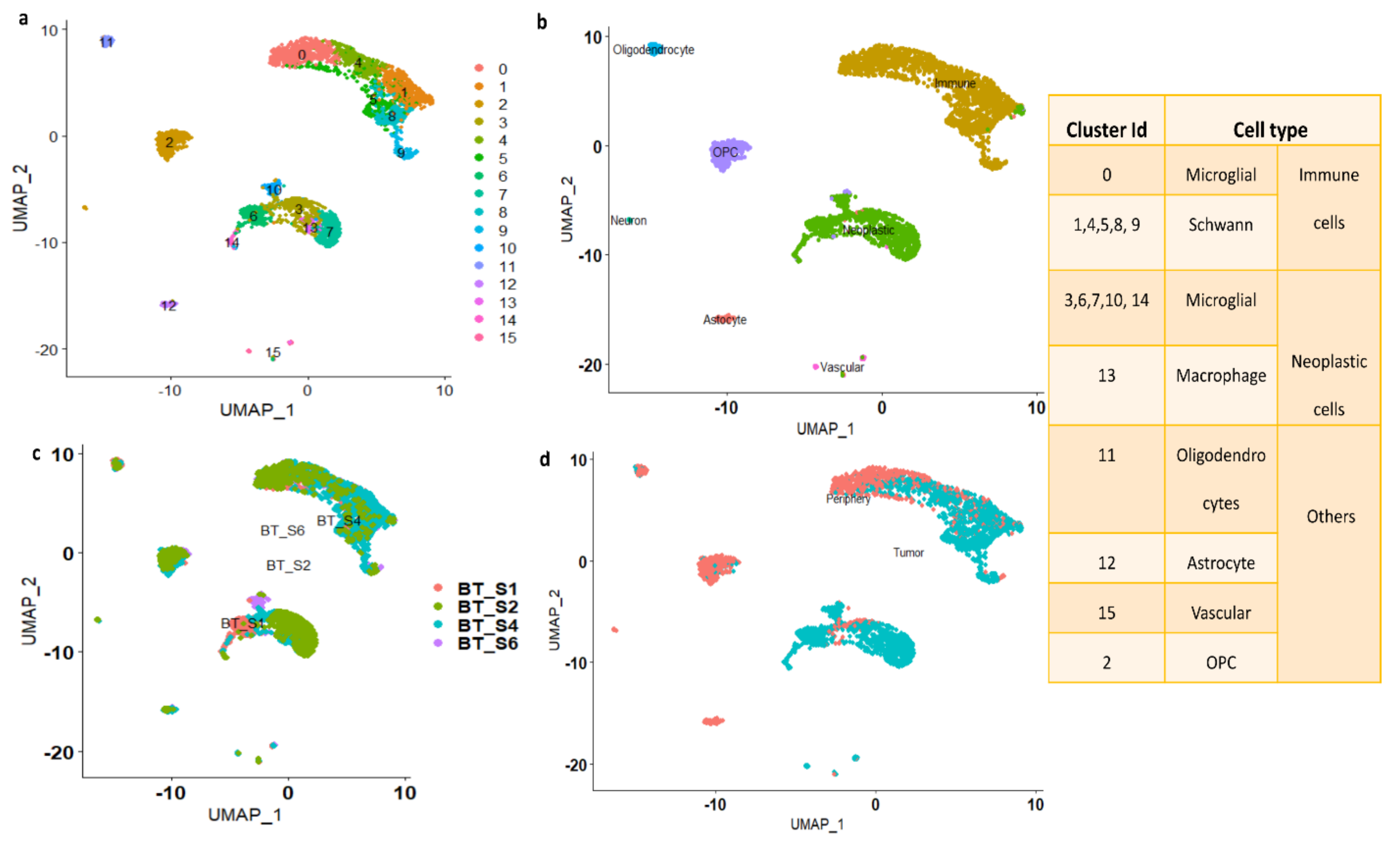
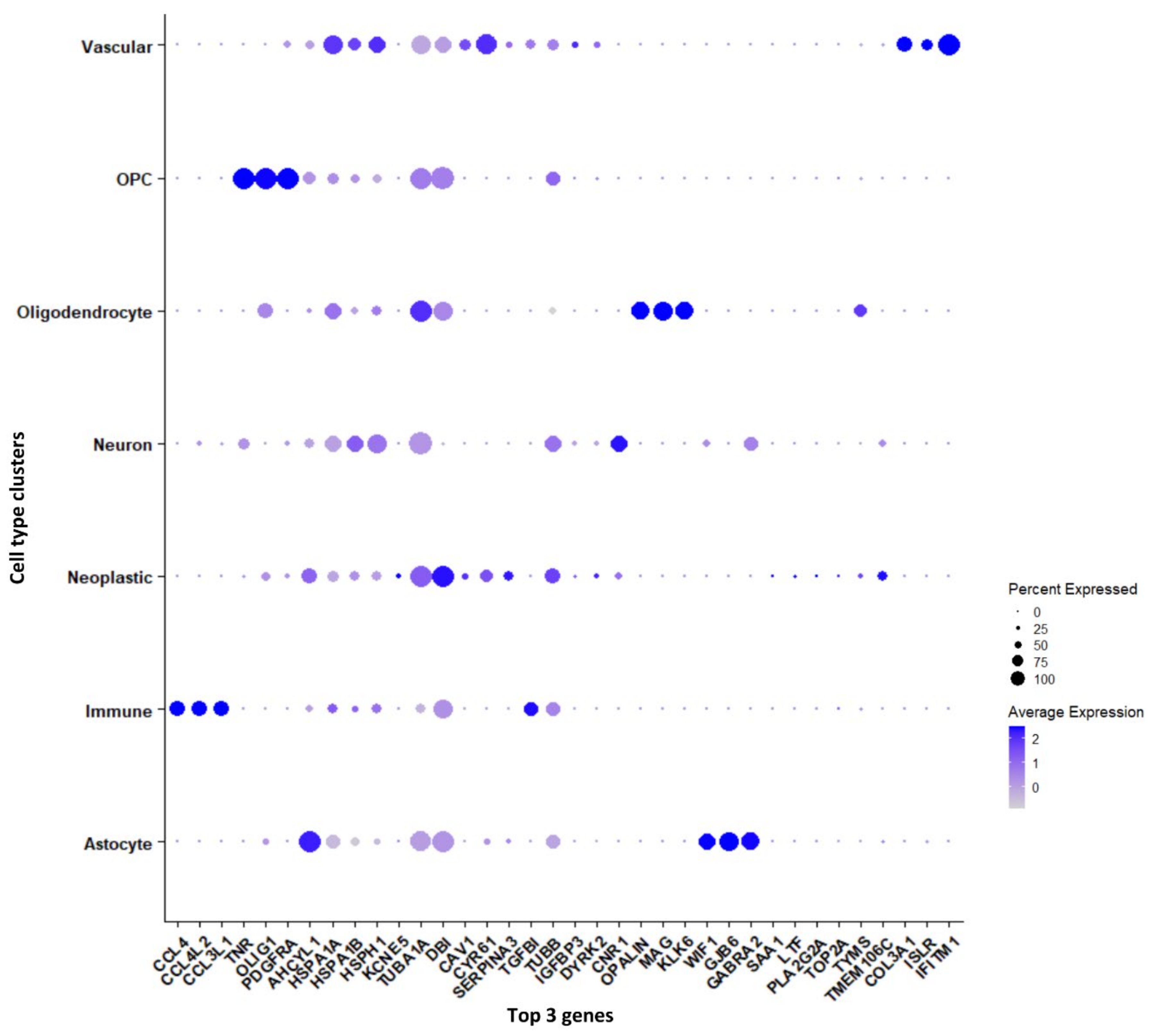
3.1.2. Marker Genes in Each Cluster
3.2. Differentially Expressed Genes (DEGs) between Tumor and Periphery Cells
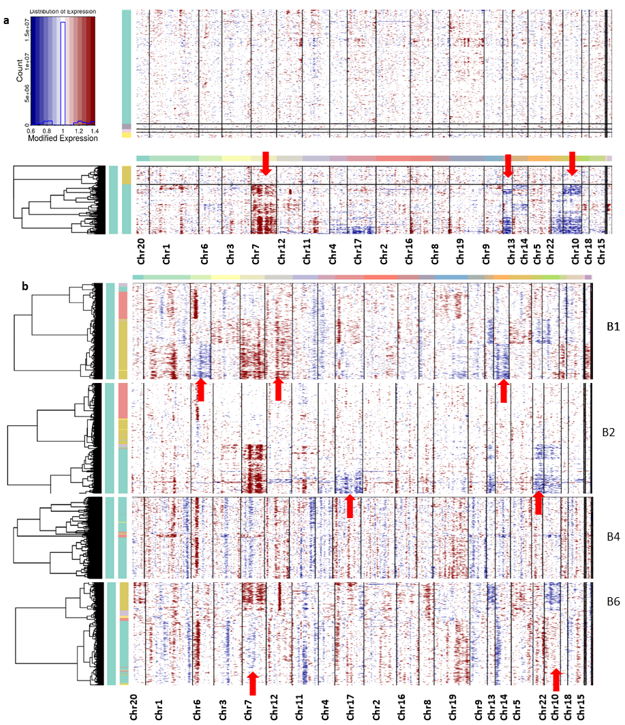
3.3. Tumor Heterogeneity with CNV Profiles
3.4. Pathway/Function Enrichment Analysis for DEGs
3.5. The Transition of Microglial Immune Cells to Neoplastic Cells: Pseudotime Analysis
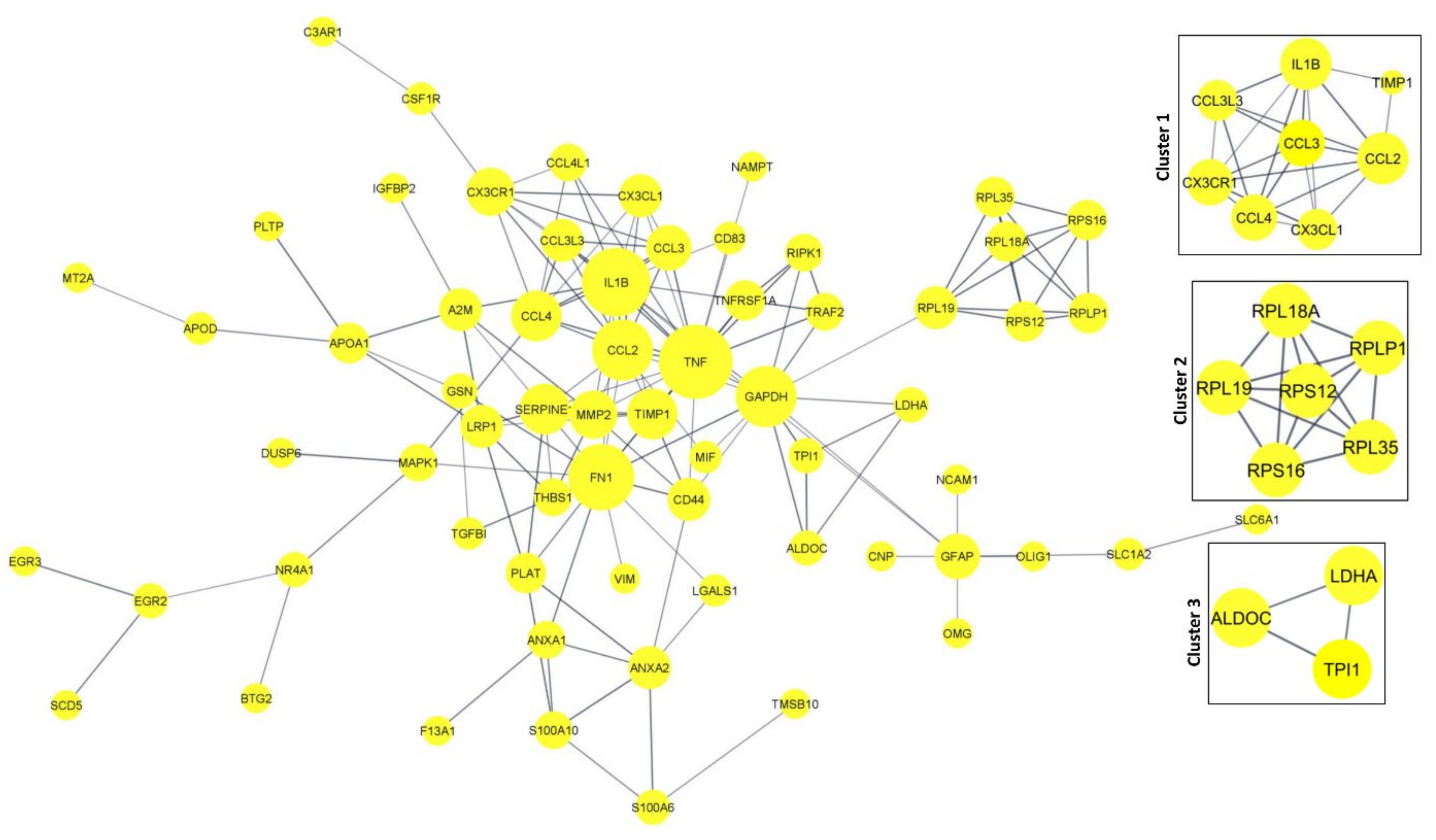
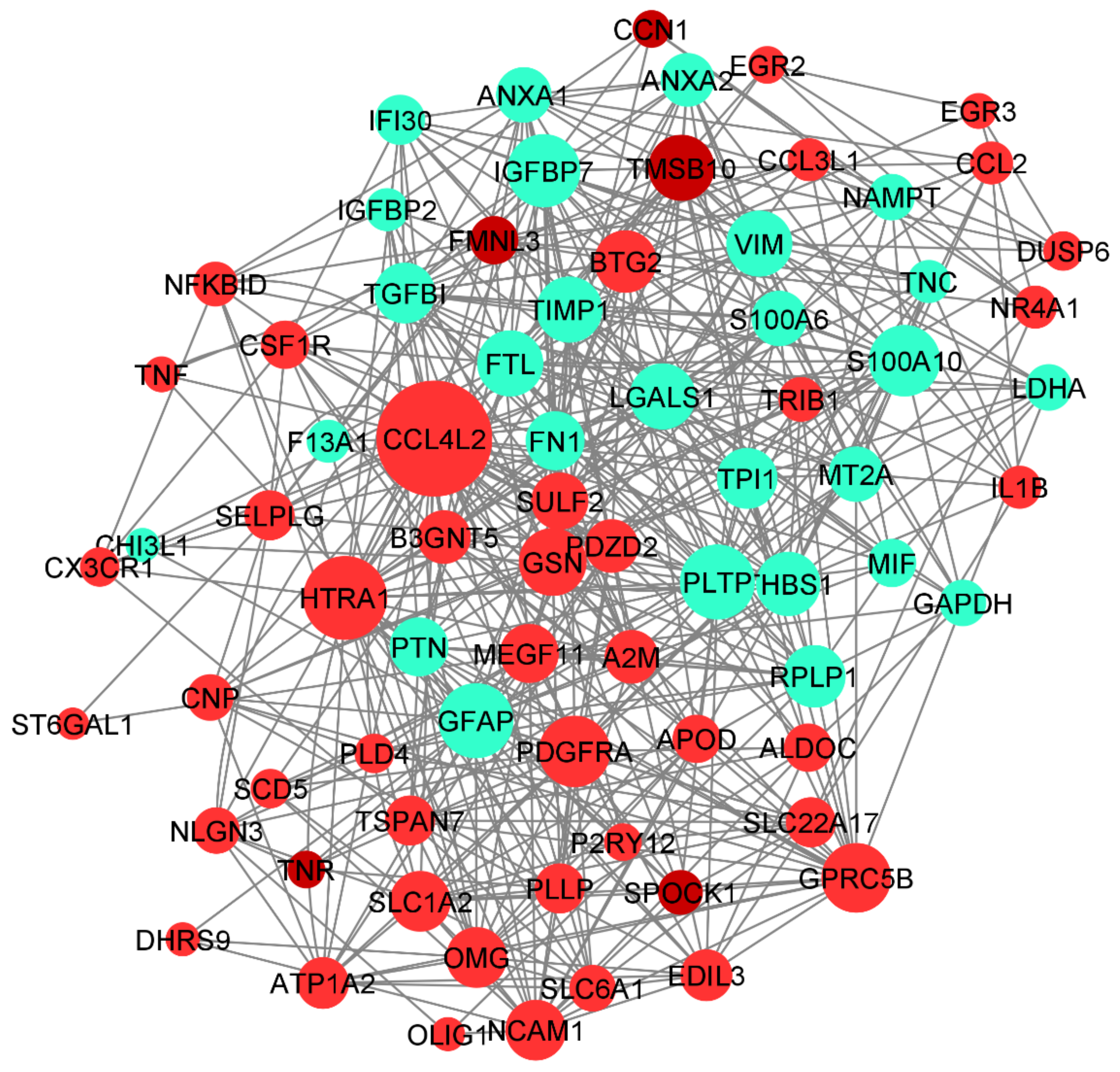
3.6. Protein–Protein Interaction Networks (PPI) and Tissue-Specific Co-Expression Networks for DEGs
3.6.1. PPI Network in DEGs
3.6.2. Tissue-Specific Co-Expression Network of DEGs
4. Discussion
Comparison with Other Related Works
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grégoire, H.; Roncali, L.; Rousseau, A.; Chérel, M.; Delneste, Y.; Jeannin, P.; Hindré, F.; Garcion, E. Targeting tumor associated macrophages to overcome conventional treatment resistance in glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Hou, C.; Chen, H.; Zong, X.; Zong, P. Genetics and epigenetics of glioblastoma: Applications and overall incidence of IDH1 mutation. Front. Oncol. 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.L.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; Malik, S.; Luu, R.; Loudig, O.; Mitchell, M.; Okafo, G.; Bhat, K.; Prideaux, B.; Eugenin, E.A. Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci. Rep. 2021, 11, 14556. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Prionisti, I.; Bühler, L.H.; Walker, P.R.; Jolivet, R.B. Harnessing microglia and macrophages for the treatment of glioblastoma. Front. Pharmacol. 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Chen, D.; Qian, Z.; Cui, D.; Gao, L.; Lou, M. Hedgehog/Gli1 signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 2017, 17, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghela, B.N.; Vaidya, F.U.; Ranjan, K.; Chhipa, A.S.; Tiwari, B.S.; Pathak, C. AGE-RAGE synergy influences programmed cell death signaling to promote cancer. Mol. Cell. Biochem. 2021, 476, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Shao, W.; Dorak, M.T.; Li, Y.; Miike, R.; Lobashevsky, E.; Wiencke, J.K.; Wrensch, M.; Kaslow, R.A.; Cobbs, C.S. Positive and negative associations of human leukocyte antigen variants with the onset and prognosis of adult glioblastoma multiforme. Cancer Epidemiol. Prev. Biomark. 2005, 14, 2040–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tickle, T.; Tirosh, I.; Georgescu, C.; Brown, M.; Haas, B. InferCNV of the Trinity CTAT Project. Klarman Cell Observatory; Broad Institute of MIT and Harvard: Cambridge, MA, USA, 2019. Available online: https://github.com/broadinstitute/inferCNV (accessed on 1 March 2021).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Alentorn, A.; Marie, Y.; Carpentier, C.; Boisselier, B.; Giry, M.; Labussiere, M.; Mokhtari, K.; Hoang-Xuan, K.; Sanson, M.; Delattre, J.Y.; et al. Prevalence, clinico-pathological value, and co-occurrence of PDGFRA abnormalities in diffuse gliomas. Neuro-oncol. 2012, 14, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Pedraza, A.M.; Cotari, J.; Liu, A.H.; Punko, D.; Kokroo, A.; Huse, J.T.; Altan-Bonnet, G.; Brennan, C.W. EGFR and PDGFRA co-expression and heterodimerization in glioblastoma tumor sphere lines. Sci. Rep. 2017, 7, 9043. [Google Scholar] [CrossRef]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgöz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.K.; Alfonso, J. Acyl-CoA-binding protein drives glioblastoma tumorigenesis by sustaining fatty acid oxidation. Cell Metab. 2019, 30, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742. [Google Scholar]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The role of hypoxia in glioblastoma invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Roche, J. The epithelial-to-mesenchymal transition in cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Kaneshiro, D.; Kobayashi, T.; Chao, S.T.; Suh, J.; Prayson, R.A. Chromosome 1p and 19q deletions in glioblastoma multiforme. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhou, W.; Ma, S.; Guan, X.; Zhang, D.; Peng, J.; Wang, X.; Yuan, L.; Li, P.; Mao, B.; et al. Identification of a Glycolysis-Related LncRNA Signature to Predict Survival in Diffuse Glioma Patients. Front. Oncol. 2021, 10, 3316. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Zupanec, N.; Urlep, Ž.; Vranič, A.; Matos, B.; Stokin, C.L.; Muyldermans, S.; Myers, M.P.; Buzdin, A.A.; Petrov, I.; et al. Differentially expressed proteins in glioblastoma multiforme identified with a nanobody-based anti-proteome approach and confirmed by OncoFinder as possible tumor-class predictive biomarker candidates. Oncotarget 2017, 8, 44141. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, K.L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T.T.; Voth, B.; Barnette, N.; Yang, I. The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Spada, S.; Tocci, A.; Di Modugno, F.; Nisticò, P. Fibronectin as a multiregulatory molecule crucial in tumor matrisome: From structural and functional features to clinical practice in oncology. J. Exp. Clin. Cancer Res. 2021, 40, 102. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Janas, M.S.; Nowakowski, R.S.; Møllgård, K. Glial cell differentiation in neuron-free and neuron-rich regions. Anat. Embryol. 1991, 184, 559–569. [Google Scholar] [CrossRef]
- Hannen, R.; Hauswald, M.; Bartsch, J.W. A rationale for targeting extracellular regulated kinases ERK1 and ERK2 in glioblastoma. J. Neuropathol. Exp. Neurol. 2017, 76, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Iwadate, Y. Epithelial-Mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.; Huang, K.; Shi, J.; Hu, Q.; Li, K.; Zhu, X. Genomics and prognosis analysis of epithelial-mesenchymal transition in glioma. Front. Oncol. 2020, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yu, X.; Sun, L.; Zheng, Y.; Chen, L.; Xu, H.; Jin, J.; Lan, Q.; Chen, C.C.; Li, M. GBP2 enhances glioblastoma invasion through Stat3/fibronectin pathway. Oncogene 2020, 39, 5042–5055. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in neurological diseases: A road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.W.E.; Lumibao, J.; Leary, S.; Sarkaria, J.N.; Steelman, A.J.; Gaskins, H.R.; Harley, B.A. Crosstalk between microglia and patient-derived glioblastoma cells inhibit invasion in a three-dimensional gelatin hydrogel model. J. Neuroinflamm. 2020, 17, 346. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-Associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/astrocytes–glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Sciumè, G.; Santoni, A.; Bernardini, G. Chemokines and glioma: Invasion and more. J. Neuroimmunol. 2010, 224, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Rzepka, Z.; Maszczyk, M.; Beberok, A.; Hermanowicz, J.M.; Pawlak, D.; Gryko, D.; Wrześniok, D. Response of human glioblastoma cells to vitamin B12 deficiency: A study using the non-toxic cobalamin antagonist. Biology 2021, 10, 69. [Google Scholar] [CrossRef]
- Chojnacki, A.; Mak, G.; Weiss, S. PDGFRα expression distinguishes GFAP-expressing neural stem cells from PDGF-responsive neural precursors in the adult periventricular area. J. Neurosci. 2011, 31, 9503–9512. [Google Scholar] [CrossRef] [Green Version]
- Del Mar Inda, M.; Fan, X.; Muñoz, J.; Perot, C.; Fauvet, D.; Danglot, G.; Palacio, A.; Madero, P.; Zazpe, I.; Portillo, E.; et al. Chromosomal abnormalities in human glioblastomas: Gain in chromosome 7p correlating with loss in chromosome 10q. Mol. Carcinog. Publ. Coop. Univ. Tex. MD Anderson Cancer Cent. 2003, 36, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Jayaram, S.; Madugundu, A.K.; Chavan, S.; Advani, J.; Pandey, A.; Thongboonkerd, V.; Sirdeshmukh, R. Chromosome-centric human proteome project: Deciphering proteins associated with glioma and neurodegenerative disorders on chromosome 12. J. Proteome Res. 2014, 13, 3178–3190. [Google Scholar] [CrossRef] [PubMed]
- Tepel, M.; Roerig, P.; Wolter, M.; Gutmann, D.H.; Perry, A.; Reifenberger, G.; Riemenschneider, M.J. Frequent promoter hypermethylation and transcriptional downregulation of the NDRG2 gene at 14q11. 2 in primary glioblastoma. Int. J. Cancer 2008, 123, 2080–2086. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Jiang, C.C.; Ho-Keung, N.; Pang, J.C.; Tong, C.Y. Chromosome 17P may harbor multiple tumor suppressor genes associated with primary glioblastoma multiforme. Chin. J. Cancer Res. 2002, 14, 60–63. [Google Scholar] [CrossRef]
- Laigle-Donadey, F.; Crinière, E.; Benouaich, A.; Lesueur, E.; Mokhtari, K.; Hoang-Xuan, K.; Sanson, M. Loss of 22q chromosome is related to glioma progression and loss of 10q. J. Neuro-oncol. 2006, 76, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.H.; Madurga, R.; García-Romero, N.; Ayuso-Sacido, Á. Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer Lett. 2022, 527, 66–79. [Google Scholar] [CrossRef]
- Kalderimis, A.; Lyne, R.; Butano, D.; Contrino, S.; Lyne, M.; Heimbach, J.; Hu, F.; Smith, R.; Štěpán, R.; Sullivan, J.; et al. InterMine: Extensive web services for modern biology. Nucleic Acids Res. 2014, 42, W468–W472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; Heiland, D.H.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.M.; Whitley, O.K.; MacLeod, G.; Cavalli, F.M.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of developmental and injury response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; He, L.; Lugano, R.; Zhang, Y.; Cao, H.; He, Q.; Chao, M.; Liu, B.; Cao, Q.; Wang, J.; et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 2021, 6, e150861. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Gene Symbol | Start Position | End Position | Description | Expression |
|---|---|---|---|---|---|
| Chr 10 | VIM | 17228241 | 17237593 | Vimentin | UP |
| HTRA1 | 1.22 × 108 | 1.23 × 108 | HtrA serine peptidase 1 | DOWN | |
| SCD * | 1 × 108 | 1 × 108 | Stearoyl-CoA desaturase | DOWN | |
| PSAP | 71816298 | 71851251 | Prosaposin | DOWN | |
| EGR2 | 62811996 | 62819167 | Early growth response 2 | DOWN | |
| PIP4K2A | 22534854 | 22714578 | Phosphatidylinositol-5-phosphate 4-kinase type 2 α | DOWN | |
| SRGN | 69057533 | 69104811 | Serglycin | - | |
| PPA1 | 70202831 | 70233429 | Inorganic pyrophosphatase 1 | DOWN | |
| OPALIN * | 96343221 | 96359002 | Oligodendrocytic myelin paranodal and inner loop protein | NI | |
| PHYHIPL * | 59175872 | 59247770 | Phytanoyl-CoA 2-hydroxylase interacting protein like | DOWN | |
| Chr 7 | ANLN | 36389806 | 36453791 | Anillin actin binding protein | UP |
| SRI * | 88205118 | 88226993 | Sorcin | UP | |
| PON2 | 95404863 | 95435329 | Paraoxonase 2 | UP | |
| ITGB8 | 20330702 | 20415754 | Integrin subunit β 8 | UP | |
| PTN | 1.37 × 108 | 1.37 × 108 | Pleiotrophin | UP | |
| CAV1 | 1.17 × 108 | 1.17 × 108 | Caveolin 1 | NI | |
| NPTX2 * | 98617297 | 98629868 | Neuronal pentraxin 2 | DOWN | |
| PTPRZ1 | 1.22 × 108 | 1.22 × 108 | Protein tyrosine phosphatase receptor type Z1 | DOWN | |
| MEST * | 1.30 × 108 | 1.31 × 108 | Mesoderm specific transcript | UP | |
| RARRES2 * | 1.50 × 108 | 1.50 × 108 | Retinoic acid receptor responder 2 | NI | |
| SEPTIN7 * | 35800932 | 35907105 | Septin 7 | NI | |
| CALD1 | 1.35 × 108 | 1.35 × 108 | Caldesmon 1 | UP | |
| GNAI1 | 80133955 | 80219402 | G protein subunit α I1 | - | |
| RAPGEF5 | 22118238 | 22357144 | Rap guanine nucleotide exchange factor 5 | UP | |
| EGFR | 55019021 | 55256620 | Epidermal growth factor receptor | DOWN | |
| IGFBP3 | 45912245 | 45921874 | Insulin-like growth factor binding protein 3 | NI | |
| COL1A2 | 94394561 | 94431232 | Collagen type I α 2 chain | DOWN | |
| GPR37 | 1.25E+08 | 1.25 × 108 | G protein-coupled receptor 37 | NI | |
| NDUFA4 | 10931951 | 10940256 | NDUFA4 mitochondrial complex associated | UP | |
| GRM3 | 86643914 | 86864884 | Glutamate metabotropic receptor 3 | NI | |
| Chr 13 | TSC22D1 | 44432143 | 44577147 | TSC22 domain family member 1 | - |
| HSPH1 * | 31134974 | 31162388 | Heat shock protein family H (Hsp110) member 1 | NI | |
| COL4A2 | 1.10E+08 | 1.11 × 108 | Collagen type IV α 2 chain | NI | |
| SLAIN1 * | 77697854 | 77764242 | SLAIN motif family member 1 | DOWN | |
| AMER2 * | 25161684 | 25172288 | APC membrane recruitment protein 2 | DOWN | |
| GPR183 * | 99294530 | 99307405 | G protein-coupled receptor 183 | DOWN | |
| PCDH9 * | 66302834 | 67230445 | Protocadherin 9 | DOWN | |
| COL4A1 | 1.10 × 108 | 1.10 × 108 | Collagen type IV α 1 chain | NI | |
| HMGB1 | 30456704 | 30617597 | High mobility group box 1 | NI | |
| Chr6 | TNF | 31575565 | 31578336 | Tumor necrosis factor | UP |
| F13A1 * | 6144084 | 6320662 | Coagulation factor XIII A chain | DOWN | |
| MYO6 | 75749203 | 75919537 | Myosin VI | DOWN | |
| AKAP12 * | 1.51 × 108 | 151358559 | A-kinase anchoring protein 12 | DOWN | |
| CD109 * | 73696203 | 73828313 | CD109 molecule | UP | |
| SLC16A10 | 1.11 × 108 | 111231194 | Solute carrier family 16-member 10 | DOWN | |
| UST * | 1.49 × 108 | 149076990 | Uronyl 2-sulfotransferase | DOWN | |
| IPCEF1 * | 1.54 × 108 | 154356803 | Interaction protein for cytohesin exchange factors 1 | UP | |
| TSPYL4 * | 1.16 × 108 | 116254075 | TSPY like 4 | DOWN | |
| SELPLG * | 1.09 × 108 | 108633894 | Selectin P ligand | UP | |
| ENO2 | 6914580 | 6923697 | Enolase 2 | NI | |
| DUSP6 | 89347235 | 89352501 | Dual specificity phosphatase 6 | UP | |
| Chr 12 | C3AR1 * | 8056844 | 8066359 | Complement C3a receptor 1 | DOWN |
| FAIM2 * | 49866896 | 49903900 | Fas apoptotic inhibitory molecule 2 | UP | |
| FMNL3 | 49636499 | 49707405 | Formin like 3 | DOWN | |
| NAV3 | 77571856 | 78213010 | Neuron navigator 3 | DOWN | |
| GPN3 | 1.1 × 108 | 110468721 | GPN-loop GTPase 3 | DOWN | |
| PRPF40B | 49622717 | 49645129 | Pre-mRNA processing factor 40 homolog B | NI | |
| LGALS3 | 55129252 | 55145430 | Complement C3a receptor 1 | UP | |
| NDRG2 | 21016763 | 21070872 | Fas apoptotic inhibitory molecule 2 | DOWN | |
| HSPA2 | 64535905 | 64543237 | Formin like 3 | DOWN | |
| RTN1 | 59595976 | 59871288 | Neuron navigator 3 | UP | |
| SLC22A17 | 23346304 | 23354991 | GPN-loop GTPase 3 | UP | |
| PLD4 | 1.05 × 108 | 104937789 | Pre-mRNA processing factor 40 homolog B | UP | |
| Chr 17 | CCL2 | 34255285 | 34257203 | C–C motif chemokine ligand 2 | UP |
| SOX9 | 72121020 | 72126416 | SRY-box transcription factor 9 | DOWN | |
| CCL3 | 36088256 | 36090143 | C–C motif chemokine ligand 3 | UP | |
| CCL4 | 36103827 | 36105614 | C–C motif chemokine ligand 4 | UP | |
| ABCC3 | 50634881 | 50692253 | ATP-binding cassette subfamily C member 3 | UP | |
| CCL4L2 | 36211063 | 36212873 | C–C motif chemokine ligand 4 like 2 | UP | |
| Chr 22 | MIF | 23894383 | 23895223 | Macrophage migration inhibitory factor | DOWN |
| LGALS1 | 37675636 | 37679802 | Galectin 1 | DOWN | |
| PDGFB | 39223359 | 39244982 | Platelet-derived growth factor subunit B | DOWN | |
| TNFRSF13C * | 41922032 | 41926806 | TNF receptor superfamily member 13C | DOWN | |
| CECR2 | 17359949 | 17558151 | CECR2 histone acetyl-lysine reader | DOWN |
| Analysis Type | Novel Genes | Total |
|---|---|---|
| Filtered DEGs | DHRS9, IPCEF1, TNR, MEGF11, EDIL3, PDZD2, ATP1A2, PDGFRA, LINC00632, AC243829.4, AC024909.2, MEG3, CHI3L1, FN1, IGFBP2, TNC, FCGBP, CYR61, F13A1, ANXA2, AC006064.4, ANXA1, CH25H and MIF-AS1 | 24 |
| CNV detection | SCD, OPALIN, PHYHIPL, PSAP, SRI, NPTX2, MEST, RARRES2, SEPTIN7, HSPH1, SLAIN1, AMER2, GPR183, PCDH9, F13A1, AKAP12, CD109, UST, IPCEF1, TSPYL4, SELPLG, C3AR1, FAIM2 and TNFRSF13C | 24 |
| Network construction | B3GNT5, SELPLG and TPI1 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yesudhas, D.; Dharshini, S.A.P.; Taguchi, Y.-h.; Gromiha, M.M. Tumor Heterogeneity and Molecular Characteristics of Glioblastoma Revealed by Single-Cell RNA-Seq Data Analysis. Genes 2022, 13, 428. https://doi.org/10.3390/genes13030428
Yesudhas D, Dharshini SAP, Taguchi Y-h, Gromiha MM. Tumor Heterogeneity and Molecular Characteristics of Glioblastoma Revealed by Single-Cell RNA-Seq Data Analysis. Genes. 2022; 13(3):428. https://doi.org/10.3390/genes13030428
Chicago/Turabian StyleYesudhas, Dhanusha, S. Akila Parvathy Dharshini, Y-h. Taguchi, and M. Michael Gromiha. 2022. "Tumor Heterogeneity and Molecular Characteristics of Glioblastoma Revealed by Single-Cell RNA-Seq Data Analysis" Genes 13, no. 3: 428. https://doi.org/10.3390/genes13030428